Cleanroom Routine Environmental Monitoring & Data Integrity with the MET ONE 3400+
Introduction
Routine Environmental Monitoring in GMP cleanrooms is a manual process and can be complex, involving thousands of sample data points per month, manual data transcription and all too frequently data gaps and integrity challenges. This paper shows how the new MET ONE 3400+ air particle counter from Beckman Coulter Life Sciences can be automated to help manage these challenges.
Cleanroom Routine Environmental Monitoring and Data Integrity Challenges
 Cleanroom environmental monitoring is done to demonstrate both that the cleanroom is operating correctly prior to the start of manufacturing and during the manufacturing period. Sample locations and frequencies are to be determined by a risk assessment. FDA CGMP1 states: “An adequate aseptic processing facility monitoring program also will assess conformance with specified clean area classifications under dynamic conditions on a routine basis.”
Cleanroom environmental monitoring is done to demonstrate both that the cleanroom is operating correctly prior to the start of manufacturing and during the manufacturing period. Sample locations and frequencies are to be determined by a risk assessment. FDA CGMP1 states: “An adequate aseptic processing facility monitoring program also will assess conformance with specified clean area classifications under dynamic conditions on a routine basis.”
However, the various GMP guidelines and International ISO standards around non-viable air particle counting in GMP cleanrooms are complex and often appear to give conflicting advice, leading to confusion and sometimes incorrect interpretation. In the case of Routine Environmental Monitoring, there is very little prescriptive advice and the onus is on the cleanroom owner to devise an appropriate monitoring plan and, in the frequently prescriptive GMP industry, the lack of direct guidance leaves users struggling to know what to do and the temptation is to either create over-burdensome monitoring programs or to try to simply use the same sample locations for routine monitoring as would be used for classification. Both are incorrect: the first because over-burdensome monitoring programs can lead to more interventions in the critical cleanroom zones than are necessary and the associated increased risk of contamination events; the second because Routine Environmental Monitoring programs should be based on a risk assessment of contamination threats to the product during the manufacturing process and the rules for Classification do not take this into account, i.e. the sampling locations for monitoring may be very different to those used in classifying the room itself once a risk assessment shows at which locations in the cleanroom the product is exposed to risk.
Carrying out a daily Routine Environmental program however still remains a manual process with potentially thousands of records taken each month, all of which typically follow a paper trail for review and approval and then are manually transcribed into electronic format. The whole process remains exposed to data errors through simple human error.
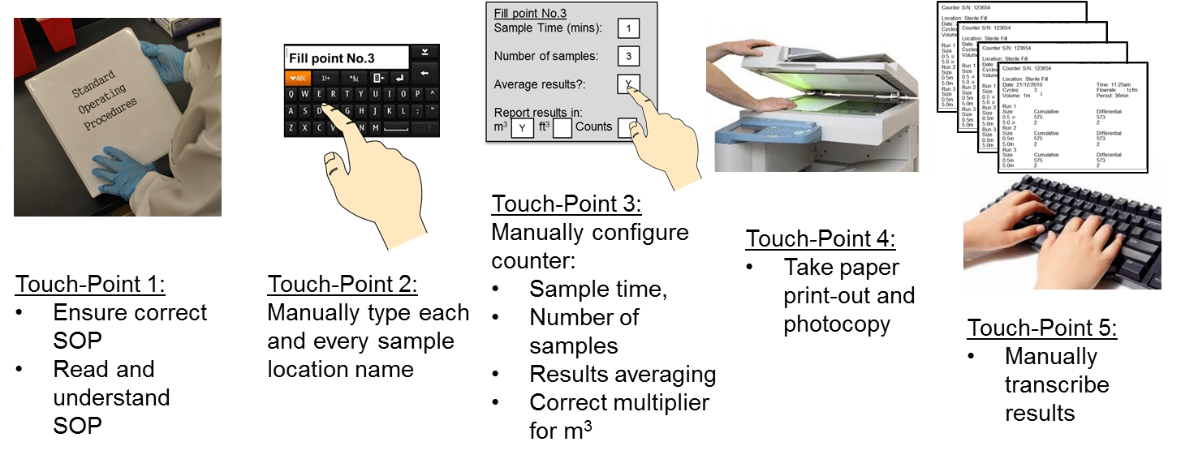
Figure 1. Routine Environmental Monitoring Programs remain subject to human error
What are the implications of data integrity for GMP cleanroom users?
The FDA’s ALCOA advice clearly targets manual configuration and data transcription practices.

The 2018 FDA Guidance on 21CFR part 11 Data Integrity Guidance for Industry2 states “In recent years, FDA has increasingly observed CGMP violations involving data integrity during CGMP inspections.” And goes on to state “These data integrity-related CGMP violations have led to numerous regulatory actions, including warning letters, import alerts, and consent decrees”
The document states “Firms should implement meaningful and effective strategies to manage their data integrity risks based on their process understanding and knowledge management of technologies”. It goes on to state that records should be “checked,” “verified,” or “reviewed”. The guidance suggests that it may be useful to ask the following questions:
- Are controls in place to ensure that data is complete?
- Are activities documented at the time of performance?
- Are activities attributable to a specific individual?
- Can only authorized individuals make changes to records?
- Is there a record of changes to data?
- Are records reviewed for accuracy, completeness, and compliance with established standards?
How can Beckman Coulter MET ONE 3400+ automated particle counter help?
Workflow features of the new MET ONE 3400+ portable air particle counter that addresses 21CFR part 11 ALCOA data records include:
Are controls in place to ensure that data is complete?
Electronic SOPs and sampling maps are uploaded and configured in the MET ONE 3400+ . SOP version control is also done in the counter. Completed sample locations turn green, allowing at a glance the user to see that the daily routine environmental monitoring program has been completed and no samples missed.
Are activities documented at the time of performance?
Electronic records of the day’s sampling activities are created contemporaneously in the MET ONE 3400+ counter itself, without the need for any external software, ensuring the activities are correctly documented at the time of performance.
Are activities attributable to a specific individual?
The MET ONE 3400+ utilizes Microsoft Active Directory for User Name and Password control and electronic signatures are attached to the electronic records that the counter creates ensuring that activities and records are attributable to a specific individual.
Can only authorized individuals make changes to records?
The MET ONE 3400+ utilizes multi-level User Name and Password controls with configurable access rights to ensure only those authorized personnel can make changes to records and any changes are recorded in the on-board Audit Trail.
Is there a record of changes to data?
Any changes are recorded in the on-board Audit Trail which can be quickly filtered and sorted to provide reports during audits.
Are records reviewed for accuracy, completeness, and compliance with established standards?
Remote review and approve via web-browser of sampling data by authorized personnel is done in the counter itself and approved electronic records with electronic signature sign-off are generated straight from the counter, avoiding paper trails and manual data transcription.
Conclusion
The MET ONE 3400+ has been designed to address the requirements of your 21CFR part 11 ALCOA data integrity requirements for GMP cleanroom routine environmental monitoring.
References
1. Food and Drug Administration. Guidance for industry. Sterile drug products produced by aseptic processing – current good manufacturing practice, 2004. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory affairs (ORA) Division of Drug Information, HFD-240 Center for Drug Evaluation and Research Food and Drug Administration 5600 Fishers Lane Rockville, MD 20857 USA
2. Food and Drug Administration. Data Integrity and Compliance With Drug CGMP Questions and Answers Guidance for Industry, 2018. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory affairs (ORA) Division of Drug Information, HFD-240 Center for Drug Evaluation and Research Food and Drug Administration 5600 Fishers Lane Rockville, MD 20857 USA
