IVD-R: Key Considerations to Validate a Flow Cytometry Assay According to ISO 15189 Requirements
Brice Ezzouaouy, Global Product Manager | Beckman Coulter Life Sciences, Marseille (France)
On this page you will
- Understand the importance of ISO 15189 for flow cytometry Laboratory Developed Tests, particularly in the context of the new EU IVD-R 2017/746 Regulation
- Learn more about the key assay performances to be verified or validated to meet ISO 15189 requirements
- Discover technical tips and concrete methodologies to meet ISO 15189 requirements
Introduction
ISO 15189:2012 Medical laboratories — Requirements for quality and competence (ISO 15189), specifies requirements for quality and competence of medical laboratories. Like other ISO standards, which aim to standardize practices globally, ISO 15189 strives to harmonize quality management system (QMS) requirements for medical laboratories performing in vitro diagnostics.
While today only selected countries across the globe have made ISO 15189 accreditation mandatory, with the upcoming EU IVD-R 2017/746 Regulation (IVD-R), accreditation becomes a requirement for all clinical laboratories across the European Union that perform Laboratory Developed Tests (LDTs). LDTs are particularly important for disciplines such as flow cytometry, as there are no ready-to-use IVD solutions commercially available for all diagnostic applications, especially in the field of hemato-oncology.
Accrediting a flow cytometry LDT according to ISO 15189 is far from straightforward, and requires careful planning. Among the many requirements covered by ISO 15189, assay performance validation can be particularly challenging, as the norm does not provide a standard procedure, or much detail, on which experiments should be performed. This whitepaper discusses some of the key assay performance characteristics requiring validation according to ISO 15189, the associated challenges specific to flow cytometry laboratories, and some key considerations for a successful journey toward ISO 15189 accreditation.
ISO 15189: overview and objectives
Based upon ISO 17025 and ISO 9001 standards, ISO 15189 is the only global standard for the accreditation of medical laboratories, providing requirements related to their competence and quality systems. The approach of a global standard aiming to harmonize and standardize quality and competence in medical laboratories across countries is unquestionably valuable. It is estimated that laboratory medicine has a role in 70 percent of clinical decisions. In the UK alone, every citizen has an average of 14 tests/year 1 performed by a laboratory medicine specialist. It is therefore critical that IVD tests are performed in controlled conditions.
The ISO 15189 standard is comprehensive, spanning the complete QMS of the laboratory. It is comprised of more than 170 articles, excluding appendixes. It encourages full involvement and use of the abilities of all employees, at all levels, to improve the organization. The goal is to ensure staff members know exactly what to do, how to do it, who is in charge of a process, and where to find all information necessary to perform their jobs.
Content of the norm is primarily divided into management (Part 4) and technical requirements (Part 5).
Table 1: Content of ISO 15189 Parts 4 and 52
|
|
|
|
While it was first released almost two decades ago (in 2003), ISO 15189 was revised in 2007 to align more closely to ISO/IEC 170253 and once more in 2012. Even though its objective was to transition from country-specific requirements to an internationally recognized standard, it has not yet been widely adopted globally. Even though it is being embraced in more countries, as of 2020 it is still not a mandatory standard in several major European countries or in the United States.
One reason it was not widely adopted across the globe is because of the complexity of its implementation. ISO 15189 is costly, time- and resource-consuming, and is not a one-time effort but a constant journey, as the principle of the norm is based on continuous improvement. The smallest labs usually don’t have the capacity to implement such a comprehensive QMS. To illustrate this point, France is a great example, as it was one of the first countries that made ISO 15189 mandatory (starting in 2011). There, the clinical laboratory landscape was completely transformed within 10 years, with the number of laboratories shrinking from 5000 in 2008 to fewer than 900 a decade later. This restructuring has been largely attributed to the necessity for smaller labs to consolidate in order to sustain accreditation requirements4.
The average cost of the accreditation process for a laboratory has been estimated at 445 000€ in the initial phase, then 145 000€ for sustainment each year thereafter5.
Another challenge for laboratories going through the ISO 15189 journey is that the norm does not specify which experiments and detailed protocols should be conducted to meet the technical requirements. It leaves room for interpretation, and it can be challenging to translate a high-level requirement into a concrete action plan.
ISO 15189: cornerstone of IVD-R compliant LDTs
The new EU Regulation for In Vitro Diagnostic Medical Devices 2017/746 (IVD-R) came into force on May 26th, 2017, with a planned, 5-year transition period. IVD-R replaces the EU Directive (98/79/EC), which had regulated in vitro diagnostic medical devices (IVDs) since 1993, as challenges emerged with the interpretation and application of that Directive, notably due to the low levels of scrutiny provided for potentially ”high risk” IVD devices.
IVD-R is a revolution for IVD manufacturers. While with the EU Directive (98/79/EC) most CE-IVD products are selfcertified, with IVD-R up to 90% of the industry now requires a notified body, and manufacturers must comply with additional (and significant) requirements related to their quality management system (QMS)—covering all applicable areas, from assay performance validation to pro-active post-market surveillance.
IVD-R not only impacts IVD manufacturers but the complete clinical diagnostics ecosystem, including end-users. This is particularly true for clinical laboratories relying on Laboratory Developed Tests (LDTs). While the IVD-R acknowledges the need for LDTs to diagnose specific pathologies, it also understands the risk associated with undercontrolled, high-risk LDTs. As the bar is being raised for manufacturers, it is logical that IVD assays developed and manufactured in laboratories are also controlled appropriately.
To perform LDTs, clinical laboratories must now meet a new set of requirements, described mostly in IVD-R article 5.5. Laboratories that perform LDTs but fail to meet article 5.5 requirements will be considered equivalent to IVD manufacturers, meaning the lab must comply with the entire regulation, (e.g., demonstrating clinical evidence, contacting a notified body and establishing post-market surveillance processes). While it is already challenging for IVD manufacturers to comply with these requirements, despite having large, cross-functional teams to support development and commercialization of assays for EU labs, it remains to be seen how many laboratories will have the capacity to perform LDTs without meeting article 5.5 requirements.
A key requirement of article 5.5 is that laboratories must comply with the EN ISO 15189 standard or, where appropriate, with national regulations, including national accreditation rules.
The figure below summarizes the typical workflow for a clinical laboratory validating an LDT vs. a CE-IVD assay, in the context of IVD-R versus IVD-D. For flow cytometry, panels built using individual CE-marked conjugated antibodies could greatly improve the validation workflow, as the assay may not be considered an LDT, and the laboratory may therefore not need to meet article 5.5 requirements. ISO 15189 compliance, or other national accreditation rules where applicable, is a cornerstone of the validation process within IVD-R, in particular for LDT performance validation.
Figure 1: IVD vs. LDT validation in the context of the IVD-D and IVD-R
 View IVDD vs IVDR infographic
View IVDD vs IVDR infographicLDT performance validation according to ISO 15189: key considerations
A key component of ISO 15189 consist in technical requirements for ensuring IVD assays demonstrate adequate performance. Method validation is potentially the most difficult and time-consuming part of the ISO 15189 journey. Technical requirements can be found in the part 5.3 Laboratory instruments, reagents and consumables and 5.5 Examination processes shown in table 1 above.
ISO 15189 provides high-level guidelines, leaving room for adapting the accreditation method depending on local specificities. This does not facilitate the task of medical laboratories, as they have to determine which procedures and experiments they must perform to meet the requirements. Some national institutions have created more concrete technical guides to support their laboratories in this journey. This is the case, for instance, in France, where the COFRAC French national accreditation body) has published concrete technical recommendations and guidance on the methodology6. A significant part of the content following in this white paper is based on COFRAC recommendations. This document is certainly not a comprehensive guide, and another methodology could certainly be acceptable, as long as it is justified and documented.
Method Validation Process Overview
IVD-R is a journey that requires careful upfront planning, in particular the method validation. The figure below highlights the key steps to verify or validate a method.
Figure 2: Major steps to verify or validate a method

If the clinical laboratory implements a method that has already been validated (e.g., CE-IVD-marked assays used according to manufacturer instructions within claimed intended use), the lab will only need to verify that the performance characteristics claimed by the manufacturer are met within this environment. If the clinical laboratory implements a method developed in-house, or modifies a validated method to better suit its needs, it will have to validate the performance characteristics. The tables below provide an example of parameters that need to be assessed, in the context of a verification or validation, for a quantitative and qualitative method.
Table 2: Parameters to be verified and/or known for a quantitative and a qualitative method6
Quantitative method
Qualitative method
Based on the results, the lab must determine the suitability of the method. If specifications defined are not met, the lab must justify their acceptance.
Repeatability and accuracy
Two key parameters to be validated are repeatability and accuracy of a system. Both are unrelated to each other. An IVD assay could be accurate but not repeatable, or vice versa, it could have a high repeatability while frequently missing the target, as illustrated in Figure 3.
Figure 3: Repeatability versus accuracy

Repeatability is assessed by analyzing the same sample multiple times—with the same operator, same reagent lot and same instrument—in a short period of time. Where possible, 2 levels of concentration, with a level close to the decision cut-off, is preferred, and 30 tests is usually ideal to ensure statistical relevance. The number of tests can vary depending on potential limitations: sample availability, cost of reagents, length of experiment, etc. The coefficient of variation is calculated based on the mean and standard deviation (=standard deviation/mean *100) and should be lower than the repeatability target defined upfront. Repeatability must be assessed for each sample type (e.g., whole blood, bone marrow). It may be necessary to assess repeatability again after a significant system intervention, such as maintenance.
Assessing accuracy typically entails a comparison to a target value, leveraging external quality controls and/or proficiency testing programs. The target value is the average value from all participants for a standardized parameter e.g., consensus value), or the average value from participants using the same method. The bias percentage will be calculated for the assay being validated as follows:
Bias % = (average from intra-lab reproducibility testing – target value)/target value x 100.
The ISO 5725 Accuracy (trueness and precision) of measurement methods and results provides more insights in its Part 4): Basic methods for the determination of the trueness of a standard measurement method.7
Intermediate Precision (within-lab reproducibility)
To assess repeatability, all parameters remain the same. To calculate intermediate precision (also called within-lab reproducibility), the same sample is analyzed multiple times under varying conditions such as: operator, time, reagent lots and instrument. A typical protocol would include 30 tests over 15 days with 2 levels. Other approaches are acceptable as long as they can be justified from a statistical perspective. Here again, the coefficient of variation is calculated based on the mean and standard deviation, and should be lower than the target defined upfront.
Linearity, limit of detection and quantification
The limit of detection is the smallest signal which can be distinguished from the background. It can be defined realizing 30 measurements on a negative control, defining the limit of detection as 3 times the standard deviation. For a qualitative method, the limit of detection will be the positive threshold.
The limit of quantification is the smallest value measured with an acceptable confidence level. It can be equal to 10 times the standard deviation from 30 negative controls measurements. Or evaluated through dilution of a control as follows:
- Perform 11 dilutions of a control (e.g., 100+0, 90+10…10+90; 0+100).
- Run each dilution 10 times and assess the coefficient of variation.
- The limit of quantification will be defined as the smallest concentration leading to a CV<10%.
An alternative approach to dilution could entail assessing different mixes of positive and negative cell lines.
The linearity between the dilutions and concentrations measured should be verified. The upper limit of linearity and limit of quantification should cover the range of concentration from samples expected to be tested in the lab.
Uncertainty and factors of variability
In any IVD assay, it is critical to define which factors may influence the result and which of these factors are nonsignificant (to be justified), and to demonstrate that other factors are controlled. Uncertainty of measurement is defined in ISO 15189 part 3.17 as “a parameter associated with the result of a measurand that characterizes the dispersion of values.”
There is not one but multiple approaches to evaluate the uncertainty of measure. For instance, it could be defined leveraging the results of internal and external quality controls, expressing the results as value ± U, where U is the uncertainty calculated with the following formula: U=√((A2+B2))
Where:
A= variance (SD2) from all internal QC
B= (mean from reproducibility study - target value)x100)/target value
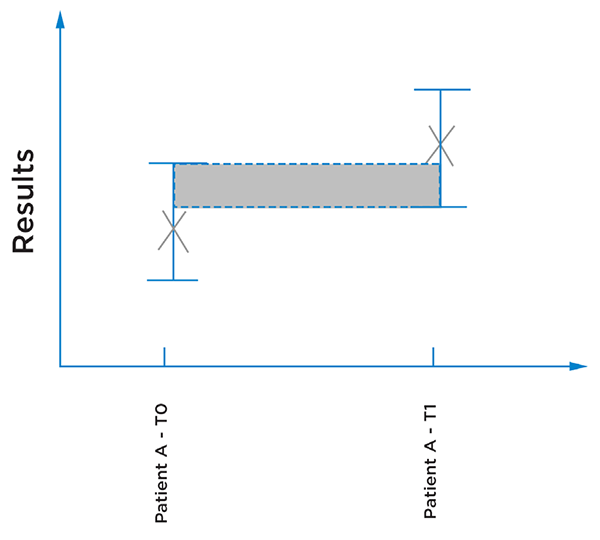
Figure 4: Illustration of a patient results from two different time points, the error bars illustrating the uncertainty of the results. Here it could not be concluded that the results are different, as it could be inherent to the imprecision of the measurement.
Reagent Stability
Definition of reagent stability is relevant only in the context of a method validation, as the laboratory can otherwise rely on the data provided by the manufacturer, as long as it follows the reagent instructions for use, and intended use and storage conditions for closed and open vials. Stability of a reagent should be demonstrated for the closed vial reagent shelf life) and open vial. This is particularly true for conjugated antibodies used in flow cytometry, as several factors could affect product performance when the vial is opened and in use (e.g., oxidation, degradation from light)8.
A classical approach to test reagent stability is to test a sample of known value (e.g., a control) from Day 1 to Day n, with a minimum of 10 replicates between Day 1 and n. The acceptable variability of the parameter measured is defined upfront (e.g. +/-10%). The limit of stability is defined as the latest value within the accepted variability range.
Figure 5: Illustration of results of a reagent stability assessment across 60 days. In this example, the reagent’s stability is 50 days, because beyond this time point results fall outside of the acceptable variability (+/- 10% in this example).

Method comparison
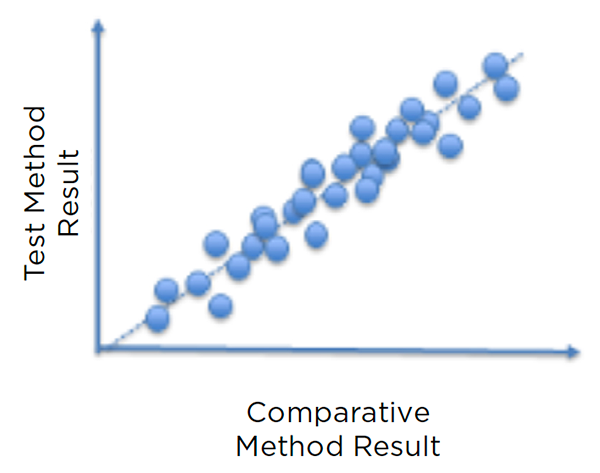
The method comparison is performed after verification of other criteria (e.g., repeatability, intra-lab reproducibility, accuracy, linearity), by running at least 30 samples, covering the range of concentrations expected to be analyzed by the lab—through both a method to be validated and the reference method—in a short period of time. Discordances are analyzed to ensure they fit within pre-established limits. If some samples fail, the potential root cause(s) must be identified (e.g., anticoagulant, specimen age) and countermeasure(s) implemented, if needed.
Figure 6: Illustration of a comparison plot showing concordance between a new method and the reference method it is validated against
Contamination across samples
Contamination across samples applies primarily to automated systems and for sensible parameters (e.g., beta-HCG). It is particularly important to assess automated sample preparation systems and washing and/or decontamination systems.
A protocol to assess contamination across samples could entail running a high-concentration sample 3 times (H1, H2, H3, mean mH), followed by a low-concentration sample 3 times (B1, B2, B3). The sequence would be repeated 5 times, and the mean of B1 and B3 calculated. Through a Student t-test, the lab can define if mB1 is different from mB3. The percentage of contamination can be calculated as: (mB1-mB3)/(mH-mB3) x 100.
The inter-reagent contamination can also be assessed when the same distribution system is used for all reagents. Here parameter A would be measured 10 times on the same sample. Then parameter A would be measured 10 times again, but alternating with parameter B. A t-test would then determine if the difference between both means is statistically different for parameter A.
Verification/Validation Dossier and continuous performance monitoring
Collecting data is great, but it is obviously useless if data are not appropriately exploited and summarized. In the context of ISO 15189 accreditation, the laboratory will create one dossier per assay. Each dossier will include, but not be limited to:
- Description of analytical process (steps, methods, elements to verify/validate)
- Risk management
- Determination of performance criteria to assess
- Determination of specifications or acceptable limits for these criteria
- Bibliography verification
- Experimental plan and implementation methodology
- Compilation and statistical analysis of data collected
- Conclusion and decision regarding the validity of the assay according to performance criteria defined
National institutions such as the COFRAC in France provide a template for these dossiers9.
The cornerstone of ISO 15189 is continuous improvement. Performance validation is not a one-time effort. Exploitation of statistical data from internal and external quality controls will enable labs to verify and confirm validity of the following performance across time:
- Intra-laboratory reproducibility must be verified periodically, particularly close to the cut-off values, and must be re-evaluated for each new lot of reagents
- Accuracy (using reference material)
- Verification (and potential adaptation) of reference values for the laboratory population
Discussion
The list of parameters to be validated and associated methodologies listed in this whitepaper are only examples, and not intended to be a comprehensive list. Each laboratory must define which parameters must be validated in the context of its IVD experiments and the norms it needs to comply with, and it must define an optimal protocol to achieve its objectives.
ISO 15189 does not specify how to address a particular requirement or clause, but encourages an effective QMS integrated across all parts of the operation, with a goal of continuous improvement.
While ISO 15189 requirements are the same for all IVD techniques, flow cytometry is certainly among the most challenging to accredit, especially flow cytometry LDTs10. Rules for accreditation have been established for highly standardized test systems. In contrast to other disciplines, such as hematology or clinical chemistry, which are highly standardized and based on automated and ready-to-use, commercially available assays, flow cytometry multicolor panels have additional layers of complexity (e.g., assay development, manual sample preparation, complex data processing/analysis).
In addition, diagnosis and monitoring of hematological malignancies is complex and dynamic. Testing protocols are constantly being reviewed in accordance with the latest opinion/data, particularly upon introduction of new markers, dyes, instruments and software that enables analysis of more parameters. Accrediting such methods through ISO 15189 requires significant effort and careful upfront planning; adding new IVD-R requirements such as the ones listed in Annex I, may limit use of LDTs in Europe only for applications where it is mandatory because no alternatives currently exist.
ISO 15189 and IVD-R are raising the bar higher for LDTs. Consequently, clinical laboratories will likely rely on LDTs only when there are no alternatives, and transition toward commercially available IVD assays, or use components already CE-IVD marked for their LDTs in accordance with the manufacturer’s instructions for use.
With IVD-R, quality and validation requirements will increase significantly for manufacturers. It seems common sense that the same IVD tests being developed in laboratories will also be more carefully controlled. This trend toward greater control of LDTs extends beyond Europe. In the U.S. for instance, the U.S. Food and Drug Administration is also considering regulating LDTs.11 The FDA has already noted it finds it problematic that a test developed by an IVD manufacturer would be regulated differently from an identical laboratory developed test.12
References
- The NHS, National Pathology Program, https://www.england.nhs.uk/wp-content/uploads/2014/02/pathol-dig-first.pdf
- International Organization for Standardization (ISO) 15189. Schneider et al. Ann Lab Med. 2017 Sep
- ISO 15189:2012 Medical laboratories - Requirements for quality and competence. Westgard QC. Pereira, P. (February 2017).
- Medical biology in the face of the evolution of health care needs. Académie Nationale de Pharmacie. 2018
- Laboratoires de biologie médicale : analyse des coûts liés à l’accréditation selon la norme EN ISO 15189. Syndicat National des Médecins Biologistes. Mai 2011. http://www.bioprat.com/modules/upload/upload/coutsaccreditation.pdf
- SH-GTA 04 technical guide, https://tools.cofrac.fr/documentation/SH-GTA-04
- ISO 5725-4:1994. Accuracy (trueness and precision) of measurement methods and results -- Part 4, https://www.iso.org/fr/standard/11836.html
- Flow cytometry and the stability of phycoerythrin-tandem dye conjugates. R. Hulspas et al. Cytometry Part A. 2009
- SH FORM 43, COFRAC. https://tools.cofrac.fr/fr/documentation/index.php?fol_id=64
- Accreditation of Flow Cytometry in Europe. Sack et al. Cytometry Part B (Clinical Cytometry) 84B:135–142 (2013)
- FDA Notification and Medical Device Reporting for Laboratory Developed Tests (LDTs), Draft Guidance Document, US FDA, 2014 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-notification-and-medical-device-reporting-laboratory-developed-tests-ldts
- Discussion Paper on Laboratory Developed Tests (LDTs), US FDA, 2017 https://www.fda.gov/media/102367/download
