17-Marker, 18-Color Human Blood Phenotyping Made Easy with Flow Cytometry
Introduction
As flow cytometry continues to develop increasing capabilities: the addition of lasers, more detectors and better signal processing, high parameter applications have begun to move out of specialty labs and into common practice. Users often find the process of establishing these high parameter applications intimidating: high-quality data requires multiple iterations of antibody-fluorochrome combinations that make up a panel and exhaustive testing to ensure sound results. Innovations in flow cytometry signal detection ease the process of panel design and data generation.
The implementation of avalanche photodiodes (APDs) for signal detection in the CytoFLEX offers two key benefits to allow for easier panel design. First, APDs are more sensitive than photomultiplier tubes (PMTs), over a wider range of the spectrum. Second, this higher photo-sensitivity results in less measurement error, which minimizes spreading. (1) Minimization of spillover spreading in high parametric experiments, allows better discrimination between dim and negative populations resulting in less critical channel selection for dim markers. Taking it one step further, ease of design can be achieved by the use of dried, unitized reagent panels such as DURAClone. The use of DURAClone IM panels as a “backbone” allows the researcher to drop in additional stains as needed, while keeping many parameters stable and pre-optimized.
Combining the innovative technologies in CytoFLEX and DURAClone allows the creation of high parameter experiments with less effort for design and set-up. In this note, we will demonstrate the ease of panel design and generation of sound data, using the CytoFLEX LX and DURAClone IM T Cell Subset panel. Moreover, with the high number of detection channels on the CytoFLEX LX, a single DURAClone IM backbone can be modified to fit multiple experimental designs and needs, allowing for quick response(s) to new questions in the lab. This paper focuses on an 18-color panel that was designed and tested quickly by using these technologies.
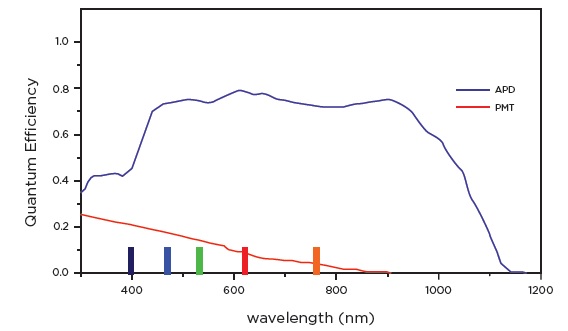
Figure 1. Comparison between PMT based and APD based systems.
Panel A. Graph representing Quantum Efficiency (QE), i.e., photon-electron conversion yield, of APDs and PMTs over the spectral range. This higher photon-electron conversion yield reduces measurement error thus facilitating higher sensitivity and resolution. Graph adapted from “A Comparison of Avalanche Photodiode and Photomultiplier Tube Detectors for Flow Cytometry” by Paul Wallace et al, 2008, Proceedings of SPI, Vol 6859. 2
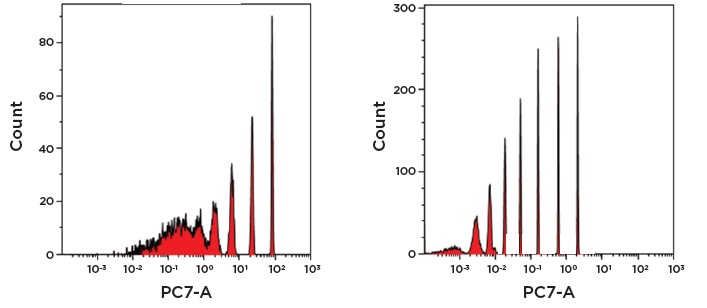
Panel B. Comparison of Spherotech 8 peak beads on PMT (left) and APD (right) shows better resolution of the dimmest beads due to increased QE, especially for emission wavelengths greater than 650 nm.

Panel C. Higher QE also contributes to less data spreading in adjacent detectors in APD based systems (right) compared to PMT based systems (left).
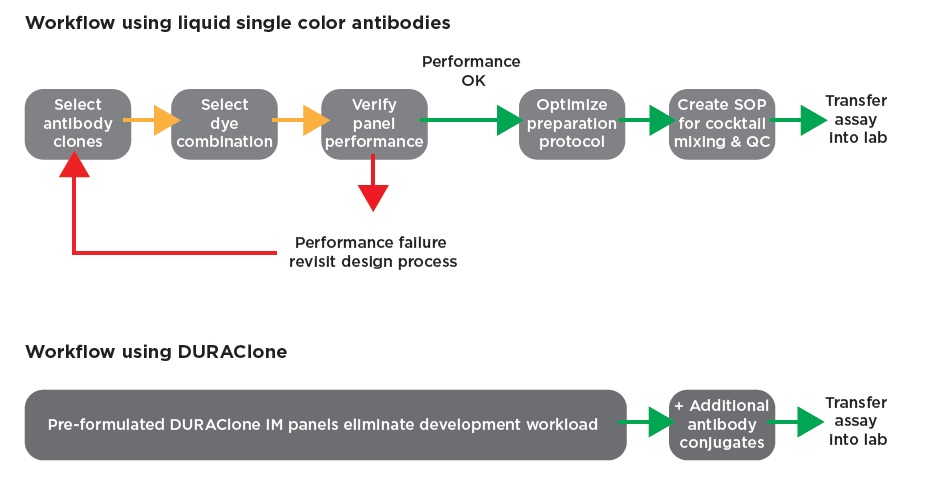
Panel D. Comparison of antibody panel design workflow with and without using DURAClone. DURAClone panels make building large panels less labor intensive by providing pre-optimized stable reagents for the backbone of the panel. Use DURAClone alone, or test and add more colors.
Materials and Methods
|
PRODUCT |
MANUFACTURER |
PART NUMBER |
|
CytoFLEX Daily QC Fluorospheres |
Beckman Coulter Life Sciences |
|
|
CytoFLEX Sheath Fluid |
Beckman Coulter Life Sciences |
|
|
CytoFLEX Daily IR QC Fluorospheres |
Beckman Coulter Life Sciences |
|
|
VersaComp Antibody Capture Beads |
Beckman Coulter Life Sciences |
|
|
VersaLyse Lysing Solution |
Beckman Coulter Life Sciences |
|
|
Dulbecco’s Phosphate Buffered Saline |
CORNING cellgro |
21-031-CV |
|
Brilliant Stain Buffer |
BD Biosciences |
566349 |
|
DURAClone IM T Cell Subsets |
Beckman Coulter Life Sciences |
|
|
CD20 BUV395 |
BD Biosciences |
563781 |
|
HLA-DR BUV661 |
BD Biosciences |
565074 |
|
CD19 BUV737 |
BD Biosciences |
564304 |
|
CCR4 BV605 |
BioLegend |
359417 |
|
CD95 BV650 |
BioLegend |
305641 |
|
CD25 BV785 |
BioLegend |
302637 |
|
CD33 PC5 |
Beckman Coulter Life Sciences |
|
|
iFluor860 (IR fixable viability dye) |
AAT Bioquest |
1408 |
|
Whole EDTA blood (24-HR post venipuncture) |
N/A |
N/A |
|
740/35 Band Pass Filter |
Beckman Coulter Life Sciences |
|
|
CytoFLEX LX UV |
Beckman Coulter Life Sciences |
TIPS FOR SUCCESS
- When using multiple Brilliant Violet or Brilliant Ultraviolet dyes, Brilliant Stain Buffer must be added to the DURAClone tube before adding these dyes to prevent dye interactions that may result in artifacts.
- Vortex DURAClone tube immediately following addition of specimen and additional antibody conjugates to ensure proper mixing of reagents.
Results

Figure 5. 17–Marker, 18–Color Panel Design. The above panel shows the marker-fluorochrome combinations used in this study. The DURAClone IM T cell Subset backbone, which consists of 10 colors, is outlined in red. The channels that were not used are shaded in gray.
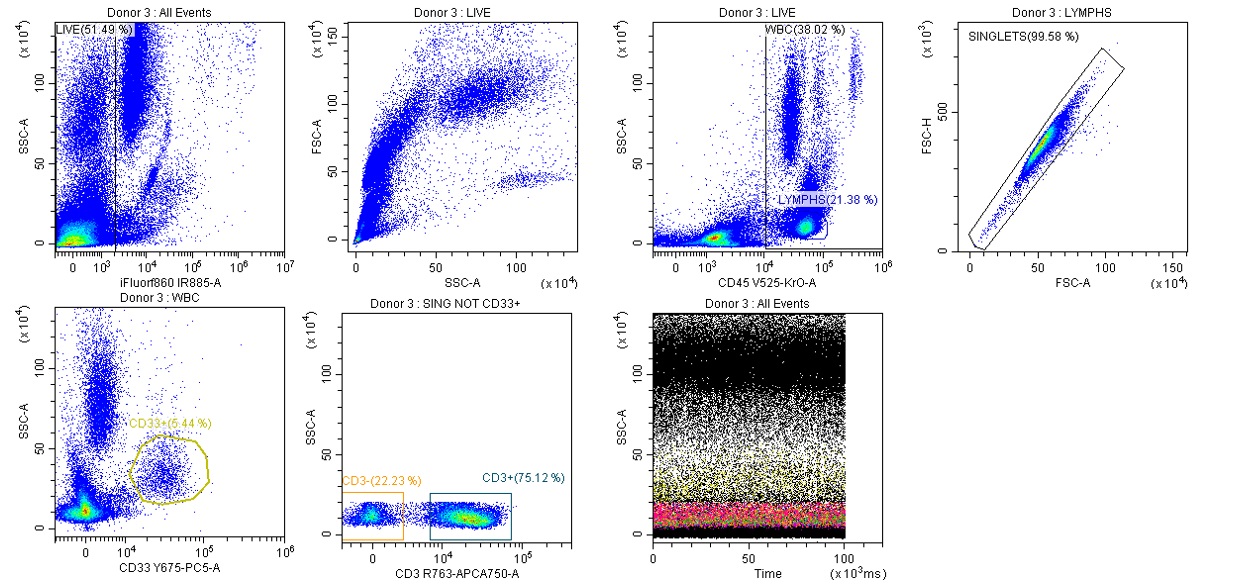
Figure 6a. High purity Gating Strategy.
CD4+ T Cells
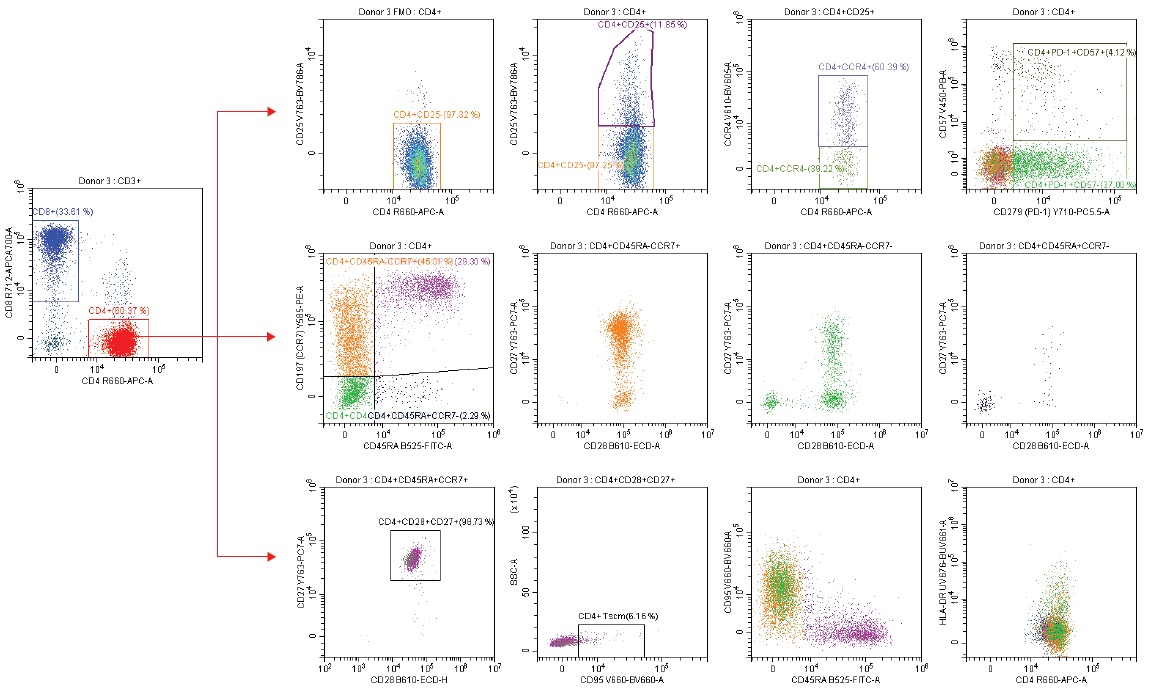
Figure 6b. CD4 T Cell Subset Analysis
CD8+ T Cells
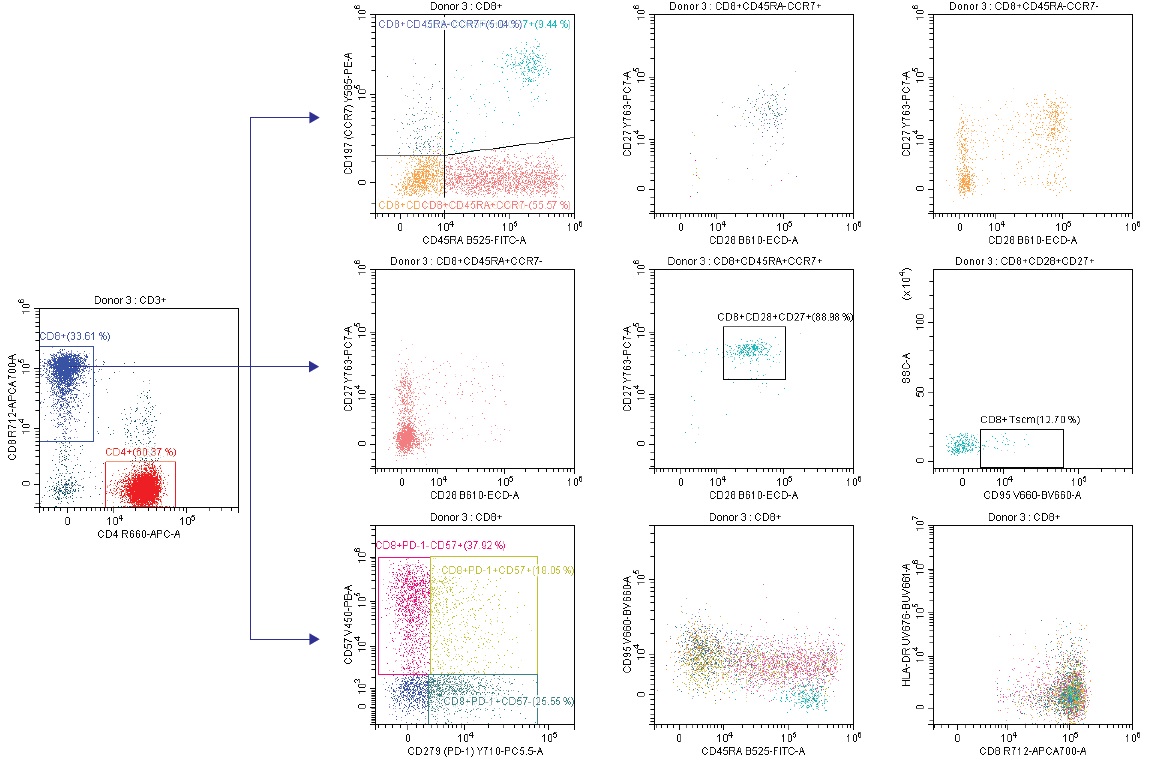
Figure 6c. CD8 T Cell Subset Analysis
B-Cells
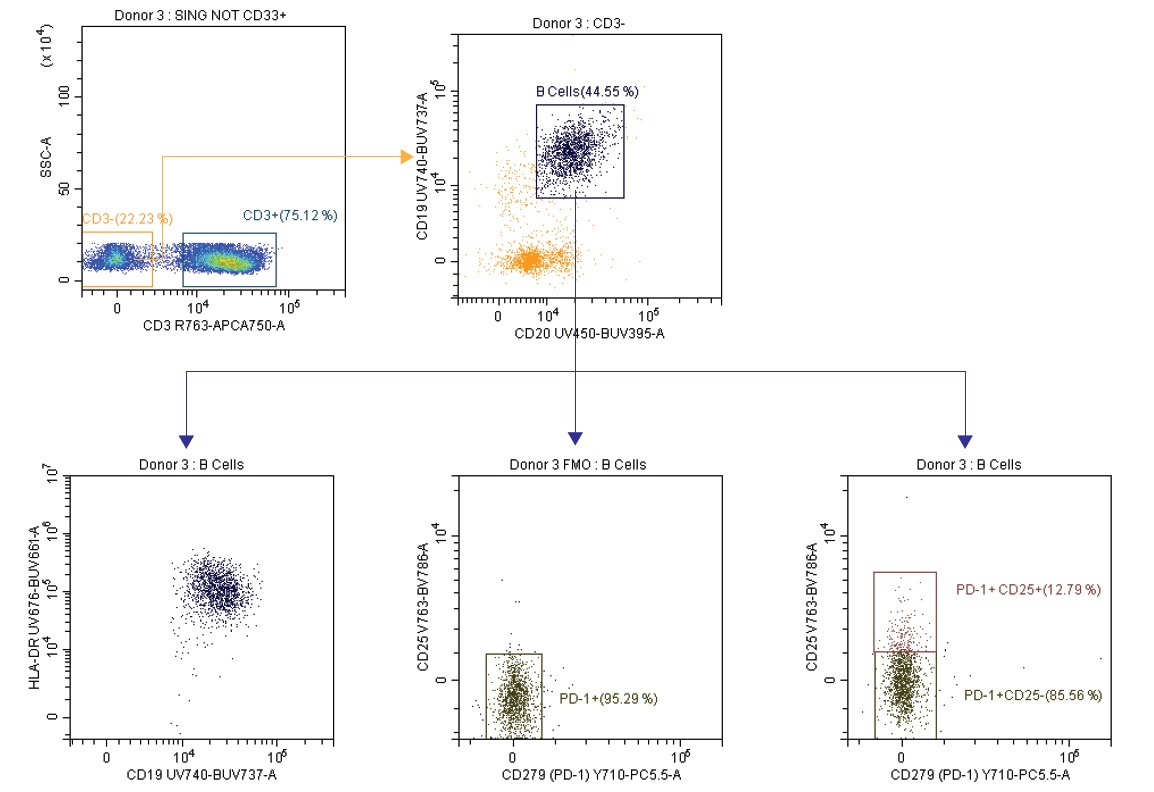
Figure 6d. B Cell Subset Analysis
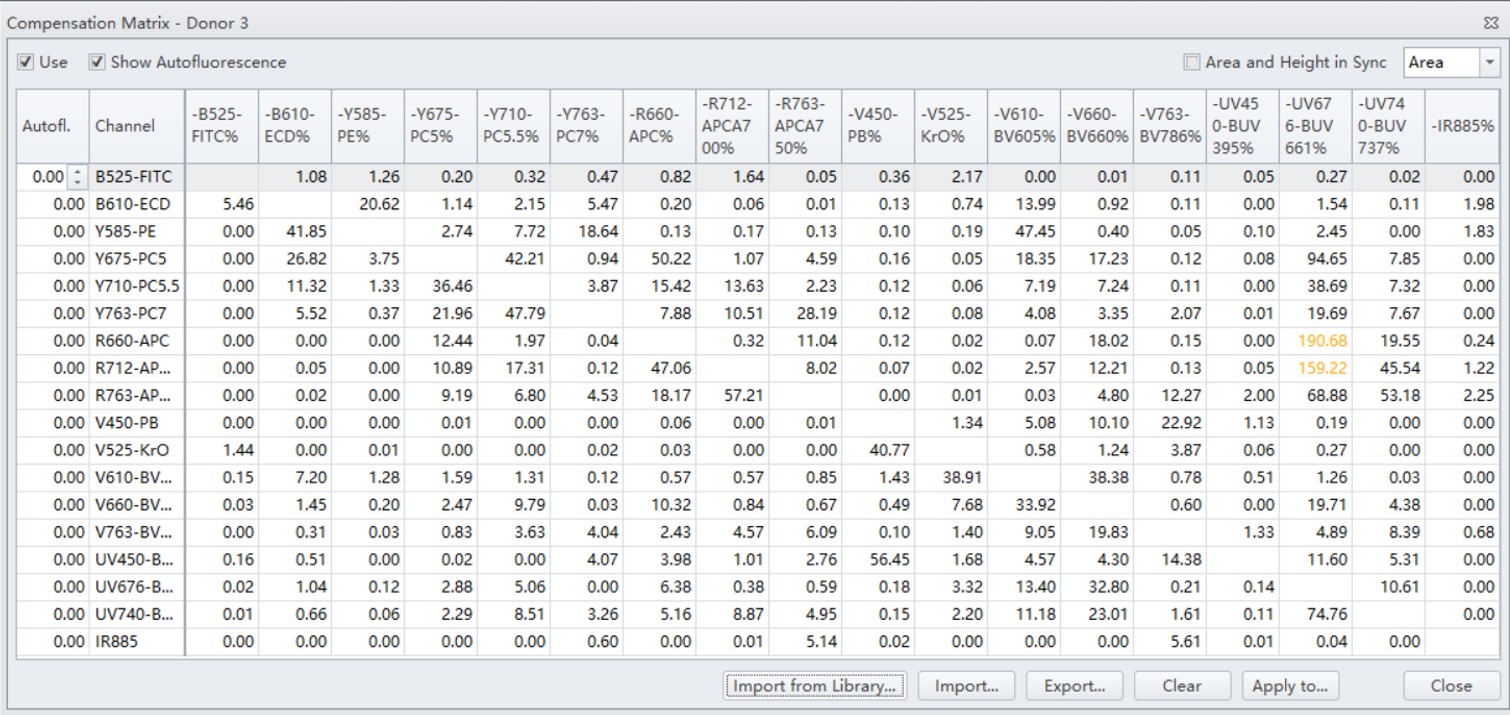
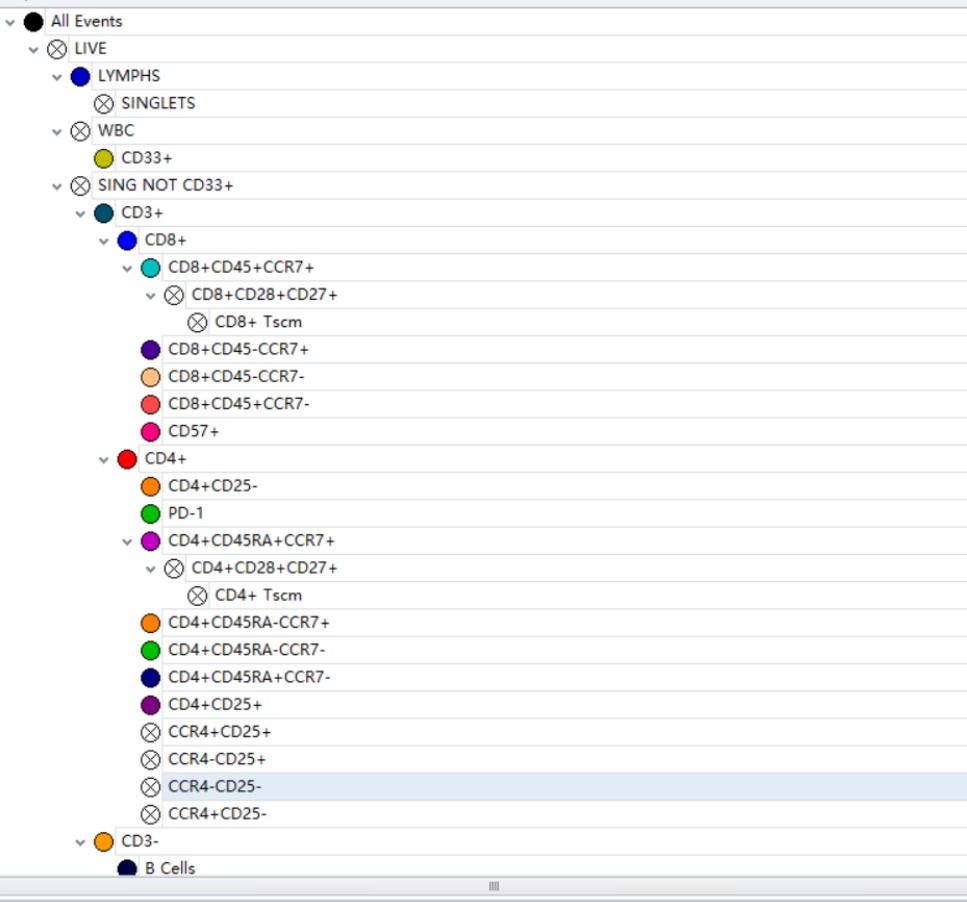
Figure 7. Compensation Matrix and Hierarchal Gating
A total of three healthy donors, 24-hours post venipuncture, were tested during this experiment. Beginning with the DURAClone IM T Cell subset, which comprises of 10 colors, as our backbone, eight markers were added to complete the panel. Marker-fluorochrome combinations adhere to the standard principles of multicolor design and also take into consideration the availability of conjugates for the desired markers.
Prior to delving into T cell analysis, a preliminary gating strategy is applied. Using the IR fixable viability dye, dead cells are excluded and the live cells gated. Live cells are then plotted as SSC vs CD45 plot, which is used to identify the White Blood Cell (WBC); a gate is drawn around the WBCs to exclude debris. The lymphocyte gate is drawn around the population that has low SSC, high CD45 fluorescence profile. Lymphocytes are further differentiated by gating out doublets and removing monocytes (CD33+) from the analysis. Lastly, we add the time plot as an internal verification of the system’s fluidics and stability during experimental runs (Figure 6a).
A total of 24 populations, but not limited to, were assessed here in the T cell subsets. To begin this analysis, we start by drawing a region on CD3+ cells falling in the lymphocyte region. This allows us to separate CD4+ and CD8+ T cells from other cell types that could express these markers, such as NK cells (CD8) and monocytes (CD4).
Taking a deeper look at the CD4 T subsets a population of CD4+CD25+ T cells comprises the Regulatory T cells. As this marker is often quite dim and therefore more difficult to gate with confidence in multicolor panels with a large potential for data spreading, we have included a Fluorescence minus One (FMO) control to increase confidence. Gating on CD4+ cells, we can also assess CCR4 expression as a function of Tregs: while the CCR4 marker plays a critical role in the homing of skin cells, a subset of CCR4+ said to control suppression of effector Regulatory T cells.3
Combining CD45RA with CCR7, we can begin to look at the various stages or pathways T cell subsets undergo during activation; i.e., naïve (CD45RA+CCR7+), central (CD45RA-CCR7+), memory, effector (CD45RA+CCR7-) and effector memory cells (CD45RA-CCR7-). Each phenotype can be further assessed by looking at the various expressions of the CD27 and CD28 co-stimulatory molecules.
Program Death Cell-1 (PD-1) is a member of the CD28 superfamily and is responsible for enhancing regulatory T cells, while impeding effector T cell function. Lastly, CD95 is responsible for cell-mediated apoptosis. CD95 is primarily expressed on memory T cells but a small portion of T cells with naïve phenotype also expresses CD95 and are considered so-called stem cell like T cells, memory type (Figure 6b).4
Taking a look at the CD8 T cell subsets, a similar approach that is used to look at CD45RA, CCR7, CD28 and CD27 marker subsets as we did with CD4. The CD8+CD57+ subset denotes terminally differentiated T cells with high cytotoxic but low proliferative capacity. CD95 expression as a function on CD45RA is once again assessed in this T cell subset. (Figure 6c)
Although not as richly stained in this panel as the T cells, B cells can also be seen (CD19+, CD20+, HLA-DR+) gating on the CD3- population. If more detail were desired in this population or other cell types, the open channels in the backbone panel should allow for ease of changes. (Figure 6d)
Conclusions
In this Application Note we illustrate the ease of using DURAClone dry unitized reagent assays as a backbone for deeper immunophenotyping panels. Beginning with a backbone and having open channels on a cytometer allows for fast results and the security of knowing that several of the parameters in the panel are pre-optimized. This method also allows for increased flexibility in the lab, as drop-in reagents can be set up to ask specific research questions.
Further, the use of DURAClone IM tubes cuts down on staff time spent dispensing reagents, and the possibility of error when pipetting multiple reagents into multiple samples tubes.
Finally, the sensitivity of APD detectors in the CytoFLEX allows for flexible high parameter panel design. Dimly expressed markers no longer require placing that marker exclusively on the high performing channels as is the case with placing CD25 BV786 on the V763 channel. Although an FMO was performed, separation between CD4+CD25- and CD4+CD25+ was clearly visible, highlighting the system’s ability to resolve dim vs negative populations.
Protocol
- Stain DURAClone compensation controls (included with DURAClone kit)
- Place one of each compensation control tube into a rack
- Add one drop each of positive and negative VersaComp beads to each tube
- Incubate for 20 minutes at room temperature, protected from light
- Add 1mL PBS+1% BSA to each tube and centrifuge at 300x g for 6 minutes
- Decant supernatant
- Vortex
- Re-suspend in 400 µL of buffer PBS+1% BSA
- Create drop-in reagent compensation controls
- Add one drop each of positive and negative VersaComp beads totube
- Incubate for 20 minutes at room temperature, protected from light
- Add 1mL buffer PBS+1% BSA to each tube and centrifuge at 300 x g for 6 minutes
- Decant supernatant
- Vortex
- Re-suspend in 400 µL of buffer PBS+1% BSA
- Prepare stained blood
- Add 50 μL of Brilliant Staining Buffer to each DURAClone tube
- Add titrated test amounts of each drop-in antibody and iFluor 860 to each DURAClone tube
- Label an additional 12 x 75 mm tube as unstained whole blood
ACQUISTION
4. Daily Startup
a. Run the CytoFLEX System Startup Program
b. Verify the detector configuration
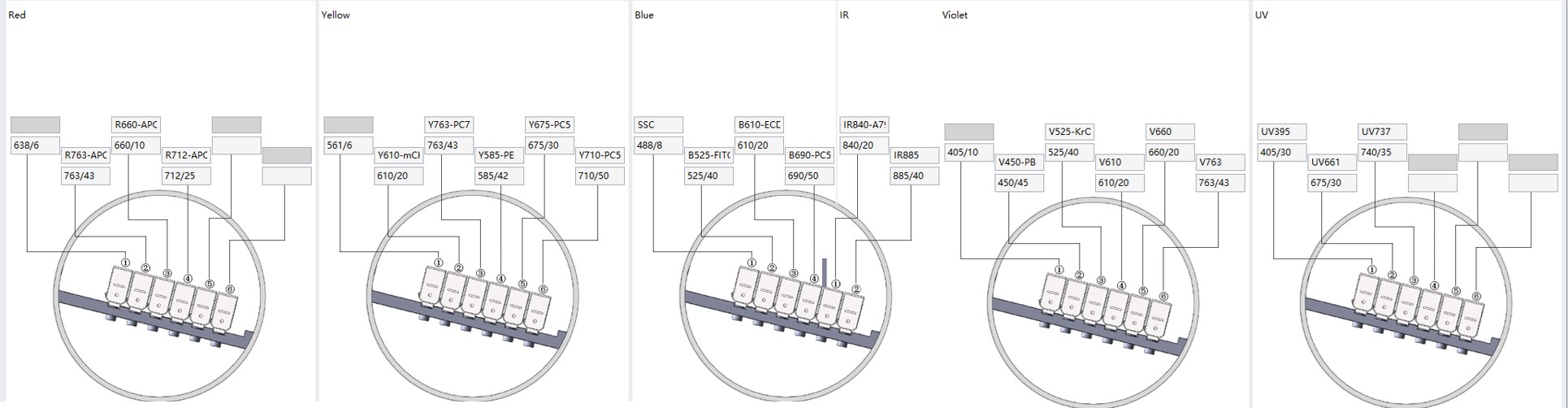
Figure 2. Detector Configuration of a CytoFLEX LX equipped with the UV laser.
c. Run the Quality Control procedure according to the user manual: “CytoFLEX Series Instructions for Use”, document number B49006xx
5. Create Compensation Experiment
a. For your DURAClone controls and drop-ins, include the lot # to allow for updating the Compensation controls when lots change
b. Select Bead in the Sample Type column to reflect your single color controls
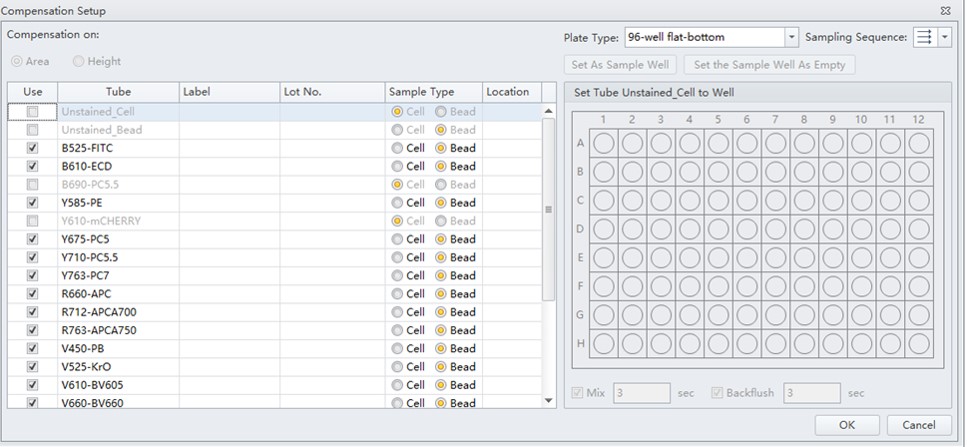
Figure 3. Compensation setup window, where the operator may select channels and sample type.
c. Uncheck using a universal negative, as VersaComp beads have a negative peak in each tube
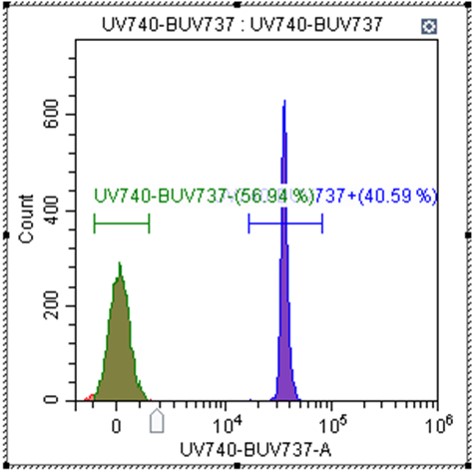
Figure 4. Example of single color run and gating.
d. Record each compensation sample
i. Move the scatter gate to contain singlet beads
ii. Use the Log-Linear slider to tighten the negative population and move the gate to contain the negative population
iii. Move the gate to contain the positive population
e. Calculate the compensation matrix and save it to the library
6. Create Experiment in CytExpert
a. Import the created compensation library and convert the matrix based on current gains
b. Create the plots
c. Record
References
1. Nguyen R, Perfetto S, Mahnke YD, et al. Quantifying spillover spreading for comparing instrument performance and aiding in multicolor panel design. Cytometry A 2013 Mar; 83 (3): 306-315. Available from URL: 2013 Feb 6; doi: 10.1002/cyto.a.22251.
2. Lawrence W, Varadi G, Entine G, et al. A Comparison of Avalanche Photodiode and Photomultiplier Tube Detectors for Flow Cytometry. Proceedings of SPIE 2008 Feb; Vol 6859. Available from URL: 2008 Feb; doi: 10.1117/12.758958.
3. Baatar D, Olkhanud P, Sumitomo K, et al. Human Peripheral Blood T Regulatory Cells (Tregs), Functionally Primed CCR4+ Tregs and Unprimed CCR4− Tregs, Regulate Effector T Cells Using FasL. J Immunol 2007 Apr 15; 178(8): 4891–4900.
4. Gattinoni L, Lugli E, Ji Y, et al. A human memory T-cell subset with stem cell-like properties. Nat Med 2011 Sep 18; 17(10): 1290–1297. Available from URL: doi:10.1038/nm.2446.
5. Van de Veen W, Stanic B, Yaman G, et al. IgG4 production is confined to human IL-10–producing regulatory B cells that suppress antigen-specific immune responses. J ALLERGY CLIN IMMUNOL 2013 APR; 131 (4).
6. Mauri C, Menon, M. The expanding family of regulatory B cells. International Immunology 2015 JUN; 27 (10): 479-486.
FLOW-4385APP11.18
