Vesicle Flow Cytometry with the CytoFLEX
John Nolan, PhD
Cellarcus Biosciences
Abstract
Extracellular Vesicles (EVs) are released by all cells and carry diverse cargo capable of eliciting functional changes in nearby or distant cells. Measurement of EVs and their cargo has broad diagnostic applicability. Manipulation of EVs and their cargo promises therapeutic utility. However, progress in the field is stifiled by characteristic limitations of existing analysis methods that introduce serious uncertainty. Common methods such as nanoparticle tracking analysis (NTA) have limited size resolution, no EV specificity, and cannot effectively measure cargo. Western blot and other methods used to measure EV cargo report total cargo in samples, not its presence on individual EVs.
Flow cytometry stands out as a potentially ideal platform for single EV cargo analysis, but in the past, instrument sensitivity and poorly standardized, non-specific assays were also insufficient for measurement of EVs. Recent advances in instrumentation, exemplified in the Beckman Coulter CytoFLEX, have resulted in significant improvement for the detection of dim signals, which has promise for EV analysis. Parallel advances in assay design have built upon improved instrumentation to enable standardized, specific, reproducible characterization of EVs. In this article we will demonstrate use of Cellarcus’ Vesicle Flow Cytometry (vFC™) assay to:
- Calibrate the instrument and determine performance across key metrics
- Sensitively detect EVs using the fluorogenic membrane probe vFRed™ and perform standardized measurement of EV concentration and size distribution
- Perform appropriately optimized, quantitative single- and multicolor EV cargo measurement
Potential and challenges for single EV analysis
Extracellular vesicles (EVs) are released by all cells and can carry molecular cargo to nearby or distant cells to affect their function. This makes EVs very interesting as mediators of intercellular signaling, as well as potential targets for development of diagnostics and therapeutics. Progress in the field to realize this potential faces a number of challenges1, most of which are limited by available methods to analyze EVs and their cargo, which tend to be non-specific, low resolution, and slow.
Review of the Minimum Information about Studies of EVs (MISEV)2 and EV-TRACK3 (transparent reporting and centralizing knowledge in EV research), reveal that the most common EV analysis methods combine ultracentrifugation (UC), which aims to enrich for small particles, with Western blotting to detect the presence of selected cargo. Often the UC pellet is subjected to nanoparticle tracking analysis (NTA) to estimate the concentration and size distribution of nanoparticles present in the sample. Interpretation of such data is challenging, owing to the facts that UC can result in significant sample loss, aggregate formation, an co-precipitation of non-EV sample contaminants4. NTA’s light scatter-based detection approach has limited size resolution and no specificity for EVs, complicating issues of UC and other enrichment methods. Bulk analysis methods such as Western blot report only the total amount of cargo in a sample, and not its presence on individual EVs. These limitations of conventional EV analysis introduce serious uncertainty into attempts to measure EVs and their cargo, including whether cargo detected in EV-containing preps is associated with EVs at all. Moreover, these methods are labor-, time-, and sample-intensive, limiting their usefulness in clinical applications.
Progress in the field urgently demands new tools that can specifically measure cargo on individual EVs with high resolution and throughput, ideally without the need for extensive sample processing.
Standardization of EV flow cytometry
Flow cytometry stands out as a potentially ideal platform for single EV cargo analysis, but several limitations have prevented widespread use5. First, most flow cytometers designed for cell analysis lack the sensitivity needed to measure very dim EVs, and struggle to resolve signals from noise. Second, most FC-based EV methods use light scatter to detect particles, an approach that lacks specificity and is challenged by very small signals compared to noise. Finally, a lack of understanding and use of the appropriate calibrators and controls renders data uninterpretable and hampers comparison of results between labs. New instruments offer enhanced sensitivity and the possibility to detect EVs and other small particles. Built-for-purpose assays and reporting leverage enhanced instrument sensitivity to enable researchers to make sensitive, specific, and quantitative measurements of individual EVs and their cargo. Detection of EVs and reproducible characterization that can be compared across labs and time requires both instruments and assays to be successful.
Recent advances in instrumentation have resulted in significant improvements in instrument sensitivity useful for EV analysis. These advances are exemplified in the Beckman Coulter CytoFLEX, which features highly efficient light collection, spectral filtering, and detectors that results in significantly improved detection of dim signals6.
Advances in assay design enable more effective EV measurement using sensitive flow cytometers. The Cellarcus Vesicle Flow Cytometry (vFC™) assay uses a fluorogenic membrane probe to selectively detect and size membrane vesicles, producing data similar to NTA or RPS, but with much greater specificity and throughput, as well as the ability to make measurements of molecular cargo. vFC™ builds on efforts to standardize EV measurement which are led by the International Society for Extracellular Vesicles (ISEV) and the International Society for the Advancement of Cytometry (ISAC). These guidelines7 aim to address the need for rigor and reproducibility in EV research and flow cytometry by developing consensus-driven recommendations for reporting on essential details. They specify a framework of controls and calibrators that are essential to facilitate comparison of methods and results across labs, a critical requirement for advancement of EV research and clinical development, and those calibrators and controls are built into vFC™ assays.
vFC&trade: Specific and quantitative EV measurement
The vFC™ Assay (Figure 1) improves upon conventional FC approaches to EV detection by using fluorescence from a membrane stain, vFRed™, rather than light scatter, to selectively detect vesicles and estimate their size. Fluorescence-triggered EV detection offers improved sensitivity and specificity compared to light scatter-based detection and simplifies sample preparation, instrument setup, and data processing. The vFC™ assay design includes essential calibration and controls required for rigorous and reproducible analysis. Performed on a sensitive flow cytometer such as the CytoFLEX6, vFC™ provides sensitive and specific measurement of EV number, size, and molecular cargo in a reproducible and scalable manner.
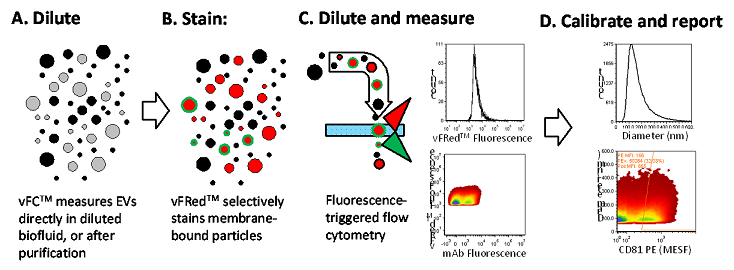
Figure 1. vFC™ assay workflow. (A) Sample (crude biofluid or enriched fractions) are diluted and (B) stained with vFRed™ and optimized fluorescence markers. (C) Stained samples are then diluted and analyzed on a sensitive flow cytometer using vFRed™ fluorescence to trigger detection. (D) Data are then calibrated and reported.
Instrument performance and calibration
Instrument performance characterization and calibration are key elements of rigorous and reproducible singe vesicle flow cytometry. Instrument optical detection performance is characterized by the efficiency of light detection and the level of background noise, which generally determines the resolution of dim particles. Instrument fluidic performance is characterized in terms of sample volumetric flow rate and linear velocity. Instrument calibration allows these measurements to be expressed in absolute units that can be compared between instruments and over time.
The vFC™ Assay Kit includes calibrators and standards for instrument performance characterization and calibration. vCal™ nanoRainbow beads, sub-micron diameter multipeak, multifluorophore hard-dyed beads, enable rapid assessment of instrument performance. Laser alignment is assessed from the CV of the brightest bead, vFRed™ resolution is assessed by the separation between the blank and dim beads, and the immunofluorescence channel intensities can be both assessed, and cross-calibrated against MESF or vCal™ calibrated antibody capture beads to provide a robust calibration particle for routine use.
Multipeak vCal™ calibrated antibody capture beads (Figure 2) enable quantitative immunofluorescence and multicolor measurements of EV cargo. Well characterized synthetic and cell-derived vesicles (Figure 2C) serve as standards for estimation of EV number, size, and molecular cargo. Together, these calibrators and standards enable quantitative measurements that can be compared across labs and over time.
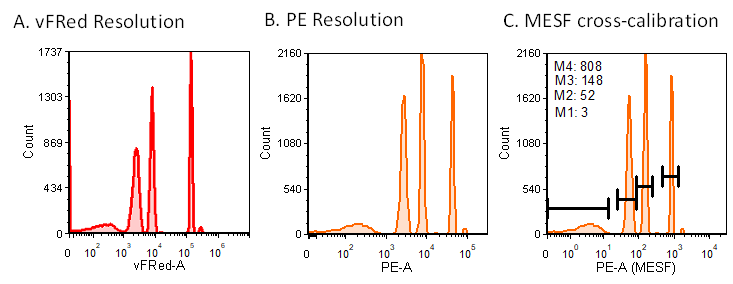
Figure 2. Instrument performance characterization and calibration. Multi-peak, multifluorophore nanoRainbow beads allow assessment of resolution in (A) vesicle detection (vFRed™) and (B) immunofluorescence (PE) channels. nanoRainbow beads can be cross-calibrated against MESF beads (C) to provide an instrument-specific calibration bead set.
vFC™ EV detection and sizing
A typical vFC™ assay workflow is presented in Figure 1. Sample is A) diluted to a particle concentration that allows single EV analysis, B) stained with a fluorogenic membrane probe (vFRed™) plus other probes (such as fluorescent antibodies), C) diluted and analyzed on the CytoFLEX, and the data is then D) loaded into the included data analysis template which calibrates and analyzes measurements to estimate EV size and cargo abundance. The entire assay takes place in a 96 well plate, can analyze EVs directly in biofluids or concentrated culture supernatants, and requires no wash steps or other sample processing.
Experiment templates prepared using the CytExpert software contain information about filter configuration, particle detection, and sample flow rate and acquisition time. For vFC™, the instrument is configured to trigger detection using vFRed™ fluorescence, and to record vFRed™ pulse width. The filters are configured to enable detection of 405 nm light scatter (VSSC) without a neutral density filter, and each well is sampled for 120 seconds at 60 uL/min (High flow rate). Use of a standardized template ensures consistent instrument operation from experiment to experiment.
Gating and data analysis
vFC™ assay data is analyzed via a standardized analysis layout that facilitates reproducible analysis and data sharing. The analysis layout includes gating to exclude fluidic anomalies (Figure 3A) and background events while selecting events with characteristic fluorescence and light scatter properties (Figure 3B).
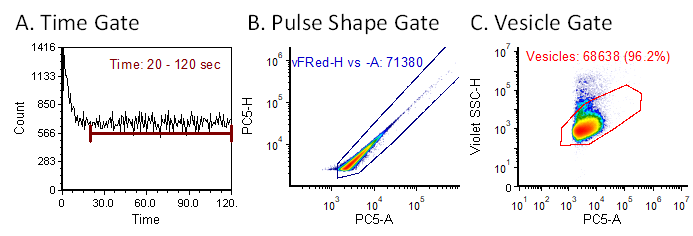
Figure 3. vFC™ vesicle gating strategy. The standardized vFCTM gating involves (A) a time gate to eliminate fluidic artifacts resulting from sample boosting, (B) gating on vFRed™ pulse shape features (height and area) to eliminate low intensity background, and (C) gating on events with characteristic light scatter and vFRed™ fluorescence.
Vesicle size measurement
In vFC™, vesicles are selectively detected via their vFRed™ fluorescence, which stains membranous particles with intensity proportional surface area. vFC™ takes advantage of these properties to estimate size from fluorescence. To illustrate, a synthetic membrane vesicle preparation, Lipo100™, containing a population of vesicles ranging from ~80-150 nm in diameter as measured by NTA or RPS (Figure 4B), is stained with vFRed™ and its fluorescence measured (Figure 4A). Plotting the Lipo100™ surface area distribution (calculated from diameter measured from orthogonal methods and assuming a spherical shape) against the fluorescence distribution (Figure 4C), we find a linear relationship (Figure 4D) and can use the slope of this line (which has units of Fluorescence unit/nm2) to estimate the Lipo100™ size from fluorescence intensity (Figure 4E,F). Thus, by using appropriate vesicle size standards, vFC™ provides EV size and concentration estimates similar to NTA or RPS, but with vesicle selectivity, which these methods lack.
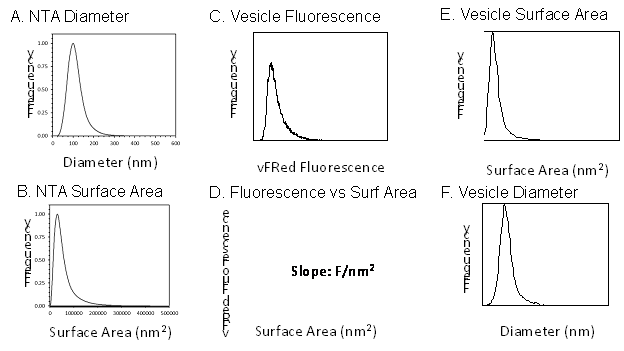
Figure 4. vFC™ size calibration. vFC™ size calibration uses a synthetic lipid vesicle (Lipo100), whose diameter (A) and surface area (B) has been estimated by an independent method such as NTA or RPS. The vFRed fluorescence distribution (C) is directly correlated with the surface area distribution allowing the vFRed intensity/surface area (D) to be calculated, and used to express vFRed fluorescence as an equivalent surface area (E) or diameter (F).
Specificity controls
The vFC™ assay protocols include the necessary controls (Figure 5) to establish assay sensitivity and EV specificity. Buffer and reagent-only controls, incorporated throughout the plate, establish the background particle and signal levels that define sensitivity (Figure 5A). A typical vFC™ run, which analyzes 100 uL of stained and diluted sample, produces <1000 background events in a buffer only control, and <2000 events in a vFRed™-only control, which results in a conservative limit of detection (LOD, background mean + 2 SD) /uL or <5000 events, or >5e4/uL, after accounting for dilution steps during sample processing.
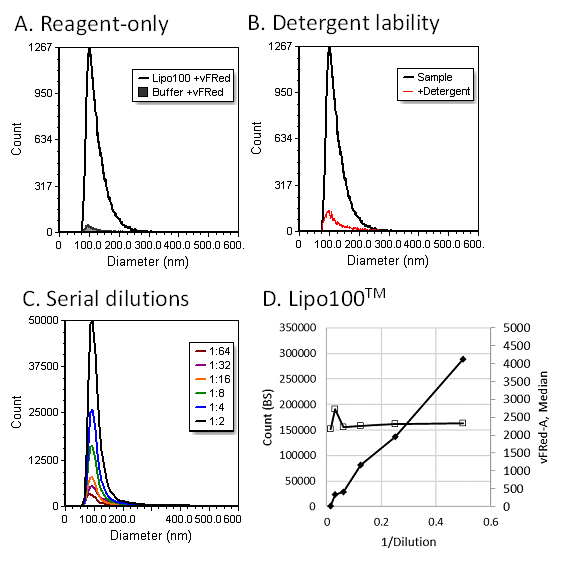
Figure 5. vFC™ Assay specificity controls. (A) Reagent-only controls allow estimation of the number of background events compared to vesicle-containing samples. (B) Detergent sensitivity testing confirms the vesicular nature of the detected events. (C) Serial dilution of sample establishes the dynamic range and sensitivity of the assay, and tests for the occurrence of coincidence (aka “swarm” detection).
A detergent control demonstrates the vesicular nature of detected events (Figure 5B). EVs and other vesicles are expected to be detergent sensitive and will be disrupted and solubilized by the addition of ~0.1% detergent. Addition of detergent can increase the background slightly, as measured in a reagent-only control, but in a sample when the events are vesicular in nature >90% of events will be eliminated. Most non-vesicular particles, including protein aggregates and lipoproteins are resistant to detergent treatment, and the presence of detergent-resistant events are an indication of the presence of such non-vesicular contaminants.
A dilution series control helps establish that the assay is measuring single particles (Figure 5C). When the number of EVs in a sample is within the linear range of the assay, the number of events in a sample decreases in proportion to dilution, but that the population distribution (and/or summary statistic such as median or mean) does not change significantly. For example, Figure 5D shows a linear decrease in number of events corresponding to dilution. The diameter distribution, however, remains constant. If the brightness of the population (and median and means) also decreases significantly, this suggest that multiple particles are being detected.
The dilution series control also establishes assay linearity and dynamic range, which typically extends from the background event counts from reagent only controls (~2000/100 uL) to ~500,000 events per 100 uL, above which point evidence of coincidence detection can often be observed. In general, assays and standards are configured to detect ~50,000 events/100 uL, with the assay working range extending a factor of 10 above and below this target.
vFC™ EV cargo measurements
One of the key limitations of other assays is inability to measure EV cargo at the single EV level. Single EV cargo measurement is required to fully understand EV heterogeneity and characterize EV subpopulations. Beyond specific and quantitative measurement of EV number and size, vFC™ coupled with the exquisite sensitivity of the CytoFLEX enables no-wash, quantitative measurement of EV cargo. EV cargo can be labeled by several means including fluorescent protein fusion expression, chemical or enzymatic labeling, or staining with fluorescently labeled antibodies with immunofluorescence being the most useful for measuring specific proteins, especially on the membrane surface. Immunofluorescence of EVs using vFC™ involves the same principles as immunofluorescence of cells, including the principles of staining optimization, specificity validation, panel design and fluorescence calibration.
vFC™ Immunofluorescence
EV surface cargo measurement depends on a calibrated instrument, validated antibodies, and optimized staining. Optimization and validation vFC™ immunofluorescence is best accomplished using appropriate calibration reagents with staining of known positive and negative samples as reference materials to demonstrate appropriate specificity and sensitivity. For example, Figure 6 shows the workflow for development of an anti-tetraspanin vTag™ antibody, the traditional positive EV marker run in vFC™ counting and sizing assays to comply with MISEV guidelines. This antibody cocktail contains multiple monoclonal antibodies with specificity to the prominent tetraspanins CD9, CD63, and CD81. The binding properties of each antibody, as well of the mixture, and optimized via antibody titiraton and appropriate controls. Antibody titration identifies the saturating concentration that provides maximal brightness while minimizing background. Specificity of staining is demonstrated using well-characterized human platelet (PLT) derived EVs as a positive control reference material, and Lipo100™ vesicle standard, which bears no protein antigen, as a negative control. An isotype control should be included to measure signal due to non-specific Fc interactions. A high signal in an isotype control necessitates pre-treatment with an Fc blocking reagent. The vTag™ anti-human TS cocktail has since been validated on EVs from dozens of sample types.
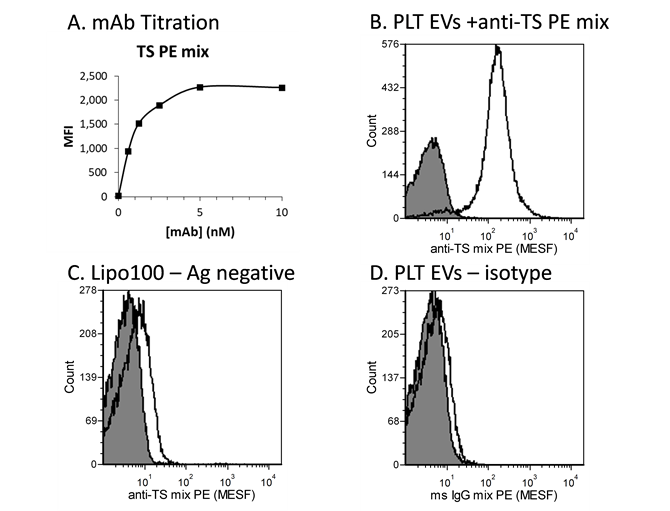
Figure 6. vFC™ Immunofluorescence. (A) Antibody titration on a positive control vesicle (PLT EVs) allows antibody staining conditions to be optimized (B) for resolution from unstained controls (shaded histogram). (C) A vesicle lacking antigen (Lipo100) can serve as a negative control. (D) An irrelevant isotype control can reveal any Fc receptor-mediated binding of antibodies.
Note: In single color staining experiments, PE is an excellent choice of a conjugate due to its brightness and wide availability. Additionally, availability of reference particles for calibration of PE fluorescence into MESF units allows the data to be expressed in absolute units, which equates with antibody molecules per vesicle for a singly labeled PE conjugate. Other colors are suitable and may be used as dictated by proper panel design.
Multicolor immunofluorescence
As with flow cytometry of cells, extending single color immunofluorescence measurements to a multicolor panel requires consideration of tenets of proper panel design. EV surface cargo is optimally measured using PE conjugates, as these are bright, widely available, and are readily calibrated using commercial reagents as discussed above. However, measurement of multiple cargos requires the design and optimization of an appropriate multicolor staining panel. Key considerations include, brightness of available conjugates, abundance of targets of interest, and expected co-expression of markers.
To illustrate, we will consider a multicolor panel to stain EVs from PLTs red blood cells (RBCs), the two most abundant cell types in blood. RBCs bear abundant CD235 (glycophorin), which is restricted to RBCs and their parent reticulocytes, and is not expressed on platelets. CD41 (alpha II integrin), on the other hand, is highly expressed on platelets and their parent megakaryocytes, but is absent from RBCs. Figure 7 shows a multicolor vFC™ assay designed to identify platelet and RBC EVs by their respective cargo. Annexin V, which binds to the procoagulant anionic phospholipid phosphatidylserine (PS) is used to stain EVs expressing surface PS. As for panel design in cell analysis, panel design in vFC™ involves consideration of antigen abundance, fluorophore brightness, spectral resolution, and availability. PE is bright and widely available, and is used here as a label for CD235, while CD41 and annexin V are labeled with BV421 and PECy5, which are also very bright and spectrally distinct, respectively. Staining antibody capture beads allows the resolution provided by the different conjugates to be assessed (a PECy5 antibody was used because annexin V is not an antibody) and the intensity to be reported in antibodies (or annexin V) bound per vesicle (ABV). Staining of EV preparations made from washed PLTs and RBCs demonstrates the specificity of CD235 and CD41 staining, as well as revealing the both PLT and RBC EVs express PS on their surface, although at ~10-fold low levels on RBC EVs compared to PLT EVs.
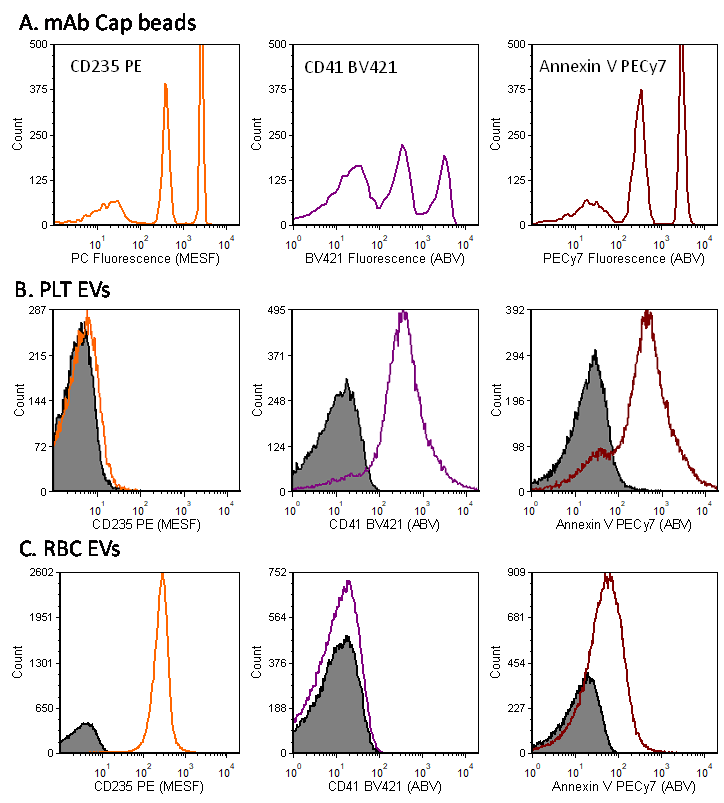
Figure 7. vFC™ multicolor immunofluorescence. (A) The brightness of fluorescence conjugates used in multi-marker panels can be measured and calibrated using vCal™ antibody capture nanobeads, which enable immunofluorescence to be expressed in terms of antibodies bound per vesicle (ABV). This enables the specificity and abundance of cell-specific and generic EV markers to be demonstrated and calibrated, for example the expression of (B) CD41 and annexin V on PLT-derived EVs and (C) CD235 and annexin V on RBC-derived EVs.
Summary and Prospects
EVs are attracting increasing interest as intercellular signaling vehicles, disease biomarkers, and therapeutic agents, but progress in all of these areas is limited by our ability to make sensitive, quantitative, and reproducible measurements of individual EVs1. Flow cytometry is a potentially powerful approach to EV measurement, but conventional flow instruments and assays lack sensitivity and specificity5. vFC™ is an assay that takes advantage of the new generation of highly sensitive flow cytometers, exemplified by the Beckman Coulter CytoFLEX6, to enable sensitive and specific measurement of EV number, size, and cargo. By combining optimized reagents and sample preparation and analysis protocols with sensitive measurement of EV fluorescence and light scatter, vFC™on the CytoFLEX enables detection of EVs to ~75 nm and detection of EV surface antibodies to ~25 molecules per vesicle. Using optimally designed and validated multicolor staining panels, we can begin to unravel the unique EV surface signatures that inform on cell of origin, possible target cells, and the potential function. These data will be key to developing informative biomarkers, therapeutic strategies, and a better understanding of the mechanisms by which these diverse entities are formed and exert their effects.
References
- Margolis, L.; Sadovsky, Y., The biology of extracellular vesicles: The known unknowns. PLOS Biology 2019, 17 (7), e3000363.
- Théry, C.; Witwer, K. W.; Aikawa, E.; Alcaraz, M. J.; Anderson, J. D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G. K.; Ayre, D. C.; Bach, J.-M.; Bachurski, D.; Baharvand, H.; Balaj, L.; Baldacchino, S.; Bauer, N. N.; Baxter, A. A.; Bebawy, M.; Beckham, C.; Bedina Zavec, A.; Benmoussa, A.; Berardi, A. C.; Bergese, P.; Bielska, E.; Blenkiron, C.; Bobis-Wozowicz, S.; Boilard, E.; Boireau, W.; Bongiovanni, A.; Borràs, F. E.; Bosch, S.; Boulanger, C. M.; Breakefield, X.; Breglio, A. M.; Brennan, M. Á.; Brigstock, D. R.; Brisson, A.; Broekman, M. L. D.; Bromberg, J. F.; Bryl-Górecka, P.; Buch, S.; Buck, A. H.; Burger, D.; Busatto, S.; Buschmann, D.; Bussolati, B.; Buzás, E. I.; Byrd, J. B.; Camussi, G.; Carter, D. R. F.; Caruso, S.; Chamley, L. W.; Chang, Y.-T.; Chaudhuri, A. D.; Chen, C.; Chen, S.; Cheng, L.; Chin, A. R.; Clayton, A.; Clerici, S. P.; Cocks, A.; Cocucci, E.; Coffey, R. J.; Cordeiro-da-Silva, A.; Couch, Y.; Coumans, F. A. W.; Coyle, B.; Crescitelli, R.; Criado, M. F.; D’Souza-Schorey, C.; Das, S.; de Candia, P.; De Santana, E. F.; De Wever, O.; del Portillo, H. A.; Demaret, T.; Deville, S.; Devitt, A.; Dhondt, B.; Di Vizio, D.; Dieterich, L. C.; Dolo, V.; Dominguez Rubio, A. P.; Dominici, M.; Dourado, M. R.; Driedonks, T. A. P.; Duarte, F. V.; Duncan, H. M.; Eichenberger, R. M.; Ekström, K.; El Andaloussi, S.; Elie-Caille, C.; Erdbrügger, U.; Falcón-Pérez, J. M.; Fatima, F.; Fish, J. E.; Flores-Bellver, M.; Försönits, A.; Frelet-Barrand, A.; Fricke, F.; Fuhrmann, G.; Gabrielsson, S.; Gámez-Valero, A.; Gardiner, C.; Gärtner, K.; Gaudin, R.; Gho, Y. S.; Giebel, B.; Gilbert, C.; Gimona, M.; Giusti, I.; Goberdhan, D. C. I.; Görgens, A.; Gorski, S. M.; Greening, D. W.; Gross, J. C.; Gualerzi, A.; Gupta, G. N.; Gustafson, D.; Handberg, A.; Haraszti, R. A.; Harrison, P.; Hegyesi, H.; Hendrix, A.; Hill, A. F.; Hochberg, F. H.; Hoffmann, K. F.; Holder, B.; Holthofer, H.; Hosseinkhani, B.; Hu, G.; Huang, Y.; Huber, V.; Hunt, S.; Ibrahim, A. G.-E.; Ikezu, T.; Inal, J. M.; Isin, M.; Ivanova, A.; Jackson, H. K.; Jacobsen, S.; Jay, S. M.; Jayachandran, M.; Jenster, G.; Jiang, L.; Johnson, S. M.; Jones, J. C.; Jong, A.; Jovanovic-Talisman, T.; Jung, S.; Kalluri, R.; Kano, S.-i.; Kaur, S.; Kawamura, Y.; Keller, E. T.; Khamari, D.; Khomyakova, E.; Khvorova, A.; Kierulf, P.; Kim, K. P.; Kislinger, T.; Klingeborn, M.; Klinke, D. J.; Kornek, M.; Kosanović, M. M.; Kovács, Á. F.; Krämer-Albers, E.-M.; Krasemann, S.; Krause, M.; Kurochkin, I. V.; Kusuma, G. D.; Kuypers, S.; Laitinen, S.; Langevin, S. M.; Languino, L. R.; Lannigan, J.; Lässer, C.; Laurent, L. C.; Lavieu, G.; Lázaro-Ibáñez, E.; Le Lay, S.; Lee, M.-S.; Lee, Y. X. F.; Lemos, D. S.; Lenassi, M.; Leszczynska, A.; Li, I. T. S.; Liao, K.; Libregts, S. F.; Ligeti, E.; Lim, R.; Lim, S. K.; Linē, A.; Linnemannstöns, K.; Llorente, A.; Lombard, C. A.; Lorenowicz, M. J.; Lörincz, Á. M.; Lötvall, J.; Lovett, J.; Lowry, M. C.; Loyer, X.; Lu, Q.; Lukomska, B.; Lunavat, T. R.; Maas, S. L. N.; Malhi, H.; Marcilla, A.; Mariani, J.; Mariscal, J.; Martens-Uzunova, E. S.; Martin-Jaular, L.; Martinez, M. C.; Martins, V. R.; Mathieu, M.; Mathivanan, S.; Maugeri, M.; McGinnis, L. K.; McVey, M. J.; Meckes, D. G.; Meehan, K. L.; Mertens, I.; Minciacchi, V. R.; Möller, A.; Møller Jørgensen, M.; Morales-Kastresana, A.; Morhayim, J.; Mullier, F.; Muraca, M.; Musante, L.; Mussack, V.; Muth, D. C.; Myburgh, K. H.; Najrana, T.; Nawaz, M.; Nazarenko, I.; Nejsum, P.; Neri, C.; Neri, T.; Nieuwland, R.; Nimrichter, L.; Nolan, J. P.; Nolte-’t Hoen, E. N. M.; Hooten, N. N.; O’Driscoll, L.; O’Grady, T.; O’Loghlen, A.; Ochiya, T.; Olivier, M.; Ortiz, A.; Ortiz, L. A.; Osteikoetxea, X.; Ostegaard, O.; Ostrowski, M.; Park, J.; Pegtel, D. M.; Peinado, H.; Perut, F.; Pfaffl, M. W.; Phinney, D. G.; Pieters, B. C. H.; Pink, R. C.; Pisetsky, D. S.; Pogge von Strandmann, E.; Polakovicova, I.; Poon, I. K. H.; Powell, B. H.; Prada, I.; Pulliam, L.; Quesenberry, P.; Radeghieri, A.; Raffai, R. L.; Raimondo, S.; Rak, J.; Ramirez, M. I.; Raposo, G.; Rayyan, M. S.; Regev-Rudzki, N.; Ricklefs, F. L.; Robbins, P. D.; Roberts, D. D.; Rodrigues, S. C.; Rohde, E.; Rome, S.; Rouschop, K. M. A.; Rughetti, A.; Russell, A. E.; Saá, P.; Sahoo, S.; Salas-Huenuleo, E.; Sánchez, C.; Saugstad, J. A.; Saul, M. J.; Schiffelers, R. M.; Schneider, R.; Schøyen, T. H.; Scott, A.; Shahaj, E.; Sharma, S.; Shatnyeva, O.; Shekari, F.; Shelke, G. V.; Shetty, A. K.; Shiba, K.; Siljander, P. R. M.; Silva, A. M.; Skowronek, A.; Snyder, O. L.; Soares, R. P.; Sódar, B. W.; Soekmadji, C.; Sotillo, J.; Stahl, P. D.; Stoorvogel, W.; Stott, S. L.; Strasser, E. F.; Swift, S.; Tahara, H.; Tewari, M.; Timms, K.; Tiwari, S.; Tixeira, R.; Tkach, M.; Toh, W. S.; Tomasini, R.; Torrecilhas, A. C.; Tosar, J. P.; Toxavidis, V.; Urbanelli, L.; Vader, P.; van Balkom, B. W. M.; van der Grein, S. G.; Van Deun, J.; van Herwijnen, M. J. C.; Van Keuren-Jensen, K.; van Niel, G.; van Royen, M. E.; van Wijnen, A. J.; Vasconcelos, M. H.; Vechetti, I. J.; Veit, T. D.; Vella, L. J.; Velot, É.; Verweij, F. J.; Vestad, B.; Viñas, J. L.; Visnovitz, T.; Vukman, K. V.; Wahlgren, J.; Watson, D. C.; Wauben, M. H. M.; Weaver, A.; Webber, J. P.; Weber, V.; Wehman, A. M.; Weiss, D. J.; Welsh, J. A.; Wendt, S.; Wheelock, A. M.; Wiener, Z.; Witte, L.; Wolfram, J.; Xagorari, A.; Xander, P.; Xu, J.; Yan, X.; Yáñez-Mó, M.; Yin, H.; Yuana, Y.; Zappulli, V.; Zarubova, J.; Žėkas, V.; Zhang, J.-y.; Zhao, Z.; Zheng, L.; Zheutlin, A. R.; Zickler, A. M.; Zimmermann, P.; Zivkovic, A. M.; Zocco, D.; Zuba-Surma, E. K., Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles 2019, 8 (1), 1535750.
- Van Deun, J.; Mestdagh, P.; Agostinis, P.; Akay, Ö.; Anand, S.; Anckaert, J.; Martinez, Z. A.; Baetens, T.; Beghein, E.; Bertier, L., EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nature Methods 2017, 14 (3), 228-232.
- Linares, R.; Tan, S.; Gounou, C.; Arraud, N.; Brisson, A. R., High-speed centrifugation induces aggregation of extracellular vesicles. Journal of Extracellular Vesicles 2015, 4.
- Nolan, J. P., Flow Cytometry of Extracellular Vesicles: Potential, Pitfalls, and Prospects. Current Protocols in Cytometry 2015, 73 (1), 13.14.1-13.14.16.
- Brittain, G. C.; Chen, Y. Q.; Martinez, E.; Tang, V. A.; Renner, T. M.; Langlois, M.-A.; Gulnik, S., A Novel Semiconductor-Based Flow Cytometer with Enhanced Light-Scatter Sensitivity for the Analysis of Biological Nanoparticles. Scientific Reports 2019, 9 (1), 16039.
- Welsh, J. A.; Van Der Pol, E.; Arkesteijn, G. J. A.; Bremer, M.; Brisson, A.; Coumans, F.; Dignat-George, F.; Duggan, E.; Ghiran, I.; Giebel, B.; Gorgens, A.; Hendrix, A.; Lacroix, R.; Lannigan, J.; Libregts, S.; Lozano-Andres, E.; Morales-Kastresana, A.; Robert, S.; De Rond, L.; Tertel, T.; Tigges, J.; De Wever, O.; Yan, X.; Nieuwland, R.; Wauben, M. H. M.; Nolan, J. P.; Jones, J. C., MIFlowCyt-EV: a framework for standardized reporting of extracellular vesicle flow cytometry experiments. J Extracell Vesicles 2020, 9 (1), 1713526.
