IVD-R 法规:根据 ISO 15189 要求验证流式细胞术检测试剂的关键考虑因素
Brice Ezzouaouy, Global Product Manager | Beckman Coulter Life Sciences, Marseille (France)
通过本文您可以:
- 了解 ISO 15189 标准对流式细胞术实验室自建检测方法的重要性,尤其是在欧盟《IVD-R 2017/746》新条例出台的背景下
- 了解更多待验证或已验证符合 ISO 15189 要求的检测试剂关键性能的相关信息
- 探索更多符合 ISO 15189 要求的技术要点和具体方法
前言
ISO 15189:2012《医学实验室质量和能力的要求》规定了医学实验室的质量与能力的要求。与其他 ISO 标准一样,ISO 15189 旨在实现医学实验室的质量管理体系(QMS)要求成为开展体外诊断业务实验室的全球通用标准。
目前,全球仅有少数国家开展 ISO 15189 标准的强制认证工作,但是新颁布的欧盟《IVD-R 2017/746 条例》(IVD-R)法规,要求欧盟国家中所有采用实验室自建检测方法(LDT)的临床实验室都应启动该体系的认证工作。LDT 对于流式细胞术等学科尤为重要,因为目前尚无适用于所有诊断应用的即用型 IVD 解决方案,尤其是在血液肿瘤学领域。
根据 ISO 15189 标准对流式细胞术 LDT 进行认证绝非易事,需要仔细规划。在 ISO 15189 认证体系中载明的众多要求中,检测试剂的性能确认尤其具有挑战性,因为该标准并未提供标准程序,或详细说明应进行哪些实验。本白皮书主要讨论一些需要根据 ISO 15189 进行确认的检测试剂关键性能特征、流式细胞术实验室面临的挑战以及成功通过 ISO 15189 认证的一些关键考虑因素。
ISO 15189: 概述与目标
规定了医学实验室能力和质量相关要求的 ISO 15189 是在 ISO 17025 和 ISO 9001 标准的基础上编制而成,是目前唯一医学实验室认可的全球通用标准。毫无疑问,其旨在实现医学实验室质量和能力统一化和标准化的全球通用标准的这一做法非常具有价值。据估计,检验医学在 70% 的临床决策中发挥着重要作用。仅就英国而言,每个公民平均每年就要通过一名检验医学专家进行 14 次检验 1 。因此,在受控条件下进行 IVD 试验至关重要。
ISO 15189 标准内容全面,涵盖完整的实验室 QMS。除附录外,含 170 多项条款。该标准鼓励全员参与,充分利用全体员工的能力改善组织执行力。其目标是确保员工确切知道要做什么、怎样做、谁负责流程以及如何找到完成工作所需的全部信息。
标准内容主要分为管理要求(第 4 部分)和技术要求(第 5 部分)。
表 1:ISO 15189第4部分和第5部分内容介绍2
|
|
|
|
ISO 15189 标准于近二十年前(2003年)首次发布,为了与 ISO/IEC 170253保持紧密一致,分别于 2007 和2012 年进行了 2 次修订。虽然其目标是希望转变为国际公认的标准,但目前尚未在全球范围内得以广泛采用。尽管越来越多的国家开始执行该标准,但截至 2020 年,欧洲几大国家或美国仍未将其视为强制性标准。
ISO 15189 此前未能实现全球通行的一个原因在于其实施的复杂性。该标准实施成本高昂,需消耗大量时间和资源,而且由于其要求持续改进,需长期运行。小规模实验室通常没有能力实施如此全面的 QMS。在这一点上,法国是典型的例证。法国是第一批强制实施 ISO 15189 (从 2011 年开始)的国家之一。10 年时间里,该国的临床实验室格局完全改变,实验室数量从 2008 年的5000 家锐减至十年后的 900 家。之所以发生这种重组情况,很大原因在于,很多小实验室必须整合才能满足认证要求4 。
实验室认证过程费用高昂,初期平均成本高达 44.5 万欧元,此后每年认证还需耗费 14.5 万欧元5。
实验室实施 ISO 15189 标准还面临另一个挑战,那就是该标准并未载明满足技术要求需进行哪些实验和落实哪些详细实验方案。该标准对此留有解释的空间,然而将高层次要求转化为具体行动计划的难度巨大。
ISO 15189: IVD-R 合规 LDT 的基石
欧盟新规《体外诊断医疗器械条例 2017/746》(IVD-R)取代欧盟指令(98/79/EC),于 2017 年 5 月 26 日生效,计划过渡期为 5年。欧盟指令(98/79/EC)从 1993 年开始成为体外诊断医疗器械监管法规,但是由于其对“高风险”IVD 器械的审查水平比较低,造成指令的解读和应用不断面临挑战,因此被淘汰。
IVD-R 法规的出台对于 IVD 制造商而言是一次革命。在欧盟指令(98/79/EC)实施期间,大部分 CE-IVD 产品可自行认证,但随着 IVD-R 的出台,目前行业内高达 90% 的 IVD 产品需依托公告机构,而且制造商必须遵守与其质量管理体系(QMS)有关的额外要求 —— 涵盖从检测试剂性能确认到积极主动上市后监督的所有适用领域。
IVD-R 法规不仅影响 IVD 制造商,对包括最终用户在内的整个临床诊断生态系统也带来冲击。对于依赖实验室自建检测方法(LDT)的临床实验室,这种影响尤为明显。虽然 IVD-R 承认 LDT 对诊断特定病理的必要性,但未受控高风险 LDT 的相关风险不容忽视。随着对制造商要求的不断提高,必然也需对实验室开发和制造的 IVD 检测试剂进行适当控制。
现在,开展 LDT 的临床实验室必须满足 IVD-R 法规第 5.5 条的新规要求。反之,将被视为等同于 IVD 制造商,这意味着这些实验室必须遵循条例的全部要求(比如提交临床证据、联系公告机构以及确定上市后监督流程)。虽然IVD 制造商拥有庞大的跨职能团队以支持欧盟实验室检测试剂的开发和商业化,但想要满足这些要求势必困难重重,究竟有多少实验室有能力在未满足第 5.5 条要求的情况下继续开展 LDT 仍有待观察。
第 5.5 条的关键要求是实验室必须遵循欧盟 ISO 15189标准,或者在适当情况下遵循国家规范,包括国家认证规则。
下图总结了临床实验室根据 IVD-R 和 IVD-D 标准对 LDT 和 CE-IVD 试剂进行确认的典型工作流。对于流式细胞术而言,使用仅经 CE 认证的偶联抗体构建样品组合可以极大改进确认工作流,原因在于该类检测试剂未被视为 LDT 的组成部分,实验室可以无需符合第 5.5 条要求。符合 ISO 15189 标准或其他国家认证规则的要求,是按照 IVD-R 进行确认的基础,特别是在 LDT 性能确认方面。
图 1:根据 IVD-D 和 IVD-R 进行 IVD 和 LDT 性能确认
 IVDD和 IVDR信息图
IVDD和 IVDR信息图根据 ISO 15189 进行 LDT 性能确认:关键考虑因素
ISO 15189 的一个关键因素包括确保 IVD 试剂盒性能达标的技术要求。方法确认可能是 ISO 15189合规之旅最艰难、最耗时的一步。相关技术要求可参考上文表 1 中提到的“5.3实验室仪器、试剂和耗材”以及“5.5 检验过程”。
虽然 ISO 15189 提供了高层次的指导方针,且允许根据当地规范调整认证方法,然而却不利于临床实验室认证工作的开展,因为它们无法明确必须执行哪些程序和实验才能符合标准要求。一些国家机构已制定出更具体的技术指南,为它们的实验室满足合规要求提供支持。例如,法国认可委员会(COFRAC)针对研究方法颁布了具体的技术建议和指南6。本白皮书中的大部分内容均以 COFRAC 建议为基础编制而成。本文当然并非全面的指南,其他方法只要合理且有章可循均可采纳。
方法确认过程概述
IVD-R 合规之旅需要提前仔细规划,尤其是在方法验证部分。下图为方法验证或确认的关键步骤。
图 2:方法验证或确认的主要步骤

如果临床实验室采用的是经验证的方法(例如,根据制造商说明在标签上载明的预期用途范围内使用带有 CE-IVD 认证标志的试剂盒),则只需验证制造商声称的性能特征是否满足此应用环境即可。如果临床实验室采用内部自建的方法,或为了更好地适应自身的要求而对已验证方法进行了改良,则其必须验证产品性能特性。下表给出定性和定量方法验证或确认过程中需要评估的参数示例。
表 2:定性和定量方法必须验证/确认的参数6
定量方法
定性方法
实验室必须根据结果确定方法的适用性。如果为满足定义的规范,实验室必须证明其是否可接受。
重复性与准确性
需要验证的两个关键参数是系统重复性和准确性。两者互不相关。IVD 检测试剂可能具有准确性但不可重复,反之亦然,它可能具有很高的重复性,但准确性却较低,如图 3 所示。
图三:重复性与准确性比较

同一操作人员在短时间内使用同一批试剂和同一台仪器对相同样品进行多次分析,即可评估重复性。如果条件允许,最好采用两种不同浓度进行对照,其中一种浓度接近临界值水平,理想试验次数为 30 次,以确保统计相关性。具体试验次数取决于潜在的限制因素:样品可用性、试剂成本、实验时长等。基于平均值和标准偏差可计算变异系数(=标准偏差/平均值*100),该值应低于预先定义的重复性目标值。必须评估每种样品种类(如全血、骨髓)的重复性。系统经重大干预(比如维护)后,可能需再次评估重复性。
评估准确性通常需要借助外部质量控制和/或熟练水平试验程序将其与目标值进行比较。目标值是指所有参与试验样品的某个标准化参数的(比如共识值)平均值,或所有参与试验样品使用同一种方法所获结果的平均值。被确认试剂的偏差百分比计算公式如下:
偏差% =(实验室内再现性测试平均值 – 目标值)/目标值 x 100
ISO 5725 测试方法与结果的准确度(正确度和精密度)在其第4部分“确定标准测试方法正确度的基本方法”中对此提供了更充分的见解。7
中间精密度(实验室内再现性)
评估重复性时,所有参数均需保持不变。计算中间精密度(又称实验室内再现性)时,需在不同条件下(比如不同的操作人员、时间、试剂批次和仪器)对同一样品进行多次分析。典型的试验方案为 15 天内使用两种不同浓度样品进行 30 次试验。其他方法只要可以从统计学角度证明其合理性,均可接受。再次说明,变异系数应基于平均值和标准偏差计算,其结果应低于预先设定的目标值。
线性度、检测限和定量限
检测限是指能够与背景区分开来的最小信号,可定义为进行 30 次阴性对照测量时,检测限为标准偏差的 3 倍。对于定性方法,检测限将为阳性阈值。
定量限是指在可接受置信水平下测得的最小值,相当于 30 次阴性对照测量标准偏差的 10 倍,或通过如下对照品稀释法进行评估:
- 将对照品稀释 11 次(例如 100+0、90+10…10+90、0+100)。
- 每种稀释液试验 10 次,评估变异系数。
- 定量限定义为 CV<10% 时的最小浓度。
另一种稀释方法需要评估不同的阳性和阴性细胞系混合样品。
稀释度和实测浓度之间的线性关系应加以验证。线性度上限和定量上限应涵盖实验室待试验样品的浓度范围。
不确定度和可变性因素
在任何 IVD 检测试剂中,确定哪些因素可能影响结果、哪些因素影响不明显(有待证明),以及证明其他因素已得到控制至关重要。ISO 15189 第 3.17部分将测量不确定度定义为“与测量结果相关的参数,用以表征合理地赋予被测量值的分散性。”
测量不确定度有多种评估方法。比如,可以利用内部和外部质量控制的结果进行定义,结果表示为数值 ± U,其中 U 为使用以下公式计算得出的不确定度: U=√((A2+B2))
其中:
A= 所有内部质量控制的方差 (SD2)
B= [(再现性研究平均值 - 目标值)x100]/目标值
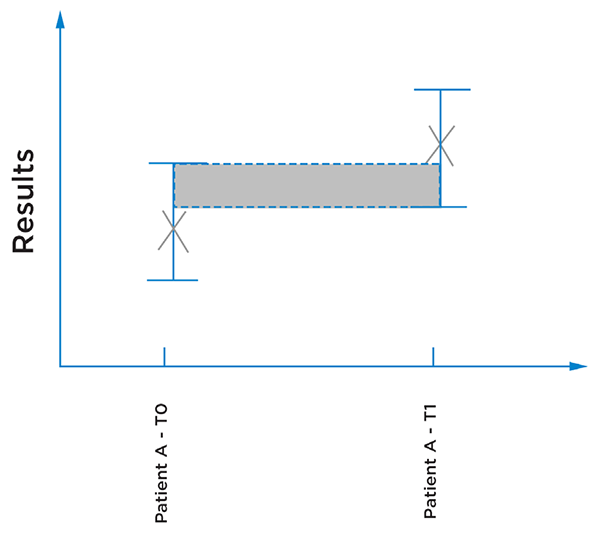
图4:表示两个不同时间点的患者检测结果,误差条表示结果的不确定度。这里无法断定结果是否不同,因为检测本身的不精确也可能导致结果不同。
试剂稳定性
试剂稳定性定义仅建立在方法确认的基础上,如果实验室按照试剂使用说明,并遵循密封和开瓶试剂的预期用途和储存条件要求,便可以依赖制造商提供的数据。试剂的稳定性,应分别对密封试剂(保质期内)以及开瓶后试剂进行验证,特别是流式细胞术所用的结合抗体更应如此,因为试剂开瓶后可能因储存条件发生变化而影响其性能(比如氧化、光降解等)8。
测试试剂稳定性的经典方法是从第 1 至 n 天连续测试具有已知值的样品(比如对照品),且在第 1 至 n 天之间至少重复测量 10 次。预先定义可接受的测量参数变异性(如 +/-10%)。稳定性极限定义为可接受变异性范围内的最新值。
图 5:展示了 60 天内试剂稳定性评估结果。在本例中,试剂稳定时间为 50 天,超过这个时间点的结果均超出可接受变异性范围(本例为 +/- 10%)。

方法比较
对其他参数指标标准(如重复性、实验室内再现性、准确度、线性度)进行验证后方法比较。方法如下:采取待确认方法和参考方法,在短时间内试验至少 30 份样品,涵盖实验室预计要分析的浓度范围。分析不一致的结果情况,确保它们未超出预先设定的限值。如果一些样品的试验结果超出限值范围,必须确定潜在的根本原因(比如抗凝剂、标本存放时间),必要时采取对策。
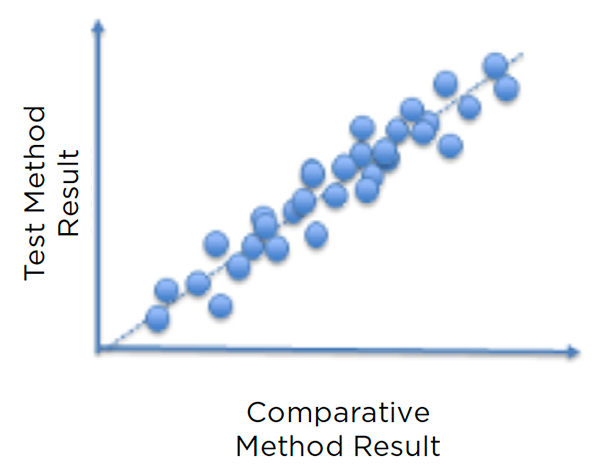
图 6:对比图显示新方法与其验证所依据参考方法的试验结果相一致
样品间污染
样品间污染主要适用于自动化系统以及敏感参数(如 β-HCG)。自动化样品制备系统和冲洗/去污系统的评估尤为重要。
评估样品间污染时,可能需要先运行高浓度样品 3 次(H1、H2、H3,平均值mH),然后再运行低浓度样品 3 次(B1、B2、B3)。上述步骤重复 5 次,计算 B1 和 B3 的平均值。通过进行 Student t 检验,实验室可确定 mB1 是否与 mB3 不一致。污染百分比可使用如下公式进行计算: (mB1-mB3)/(mH-mB3) x 100。
如果所有试剂使用同一个分配系统,也可以评估试剂间污染。使用同一样品测量 10 次参数 A。然后参数 A 和参数 B 交替测量,各运行 10 次。对于参数 A 而言,t 检验将确定两个平均值之间的差异是否具有统计学意义。
验证/确认档案与持续性能监测
数据收集很重要,但是如果不加以适当利用和总结,收集的数据显然毫无意义。依据 ISO 15189 认证标准,实验室需为每份检测试剂创建一份档案。档案内容包括但不限于:
- 分析过程描述(验证/确认的步骤、方法、要素)
- 风险管理
- 待评估性能标准的确定
- 这些标准规格或可接受限值的确定
- 参考文献验证
- 实验计划及实施方法
- 已收集数据整理和统计分析
- 根据定义的性能标准对检测试剂的有效性作出的结论和决定
法国 COFRAC 等国家机构为此类档案的编写提供了模板9 。
ISO 15189 强调持续改进。性能确认无法一蹴而就。根据内部和外部质量控制统计数据,实验室可以验证和确认以下性能随时间变化的有效性:
- 实验室内再现性,必须定期验证,尤其是接近临界值的再现性,而且必须重新评估每批新试剂
- 准确度(使用参比材料)
- 实验室群体参比值验证(以及潜在调整)
讨论
本白皮书列出的待确认参数和相关方法仅供参考,内容并不全面。每个实验室须根据其开展的 IVD 实验确定待验证参数以及需遵循的规范,并拟定达成目标的最佳方案。
ISO 15189 未明确如何处理特定的要求和条款,但鼓励以持续改进为目标,将有效的 QMS 整合至标准运行的各个环节。
虽然 ISO 15189 标准对所有 IVD 技术的要求均一致,但流式细胞术(尤其是流式细胞术 LDT)的认证工作无疑是最具挑战性的项目之一10。高度标准化试验系统已经制定了认证规则。与已实现高度标准化且基于市售自动化即用型试剂盒开展试验的其他学科(例如血液学或临床化学)相比,流式细胞术多色组合试剂更为复杂(例如,检测试剂开发、手动样品制备以及复杂的数据处理/分析等)。
此外,血液系统恶性肿瘤的诊断和监测具有复杂性和动态性。根据最新的意见/数据显示,特别是在引入可分析更多参数的新标记物、染料、仪器和软件后,测试方案需不断进行审查。根据 ISO 15189 标准认证这些方法需要提前仔细规划并付出巨大的努力;附件 I 所载的 IVD-R 要求,可能会限制 LDT在欧洲的应用,造成因没有其他选择而只能强制使用 LDT 的应用窘境。
ISO 15189 和 IVD-R 抬高了 LDT 的门槛。因此,临床实验室可能只有在没有其他选择时才会考虑 LDT,并转而寻求市售 IVD 试剂盒,或根据制造商使用说明在其 LDT 中使用带 CE-IVD 认证标志的试验耗材。
随着 IVD-R 的出台,针对 IVD 制造商的质量和确认要求将显著增加。似乎大家都认为,实验室正在研发的 IVD 检测方法也将受到更严格的管控。这种加大 LDT 监管力度的趋势不仅限于欧洲。以美国为例,美国食品药品监督管理局(FDA)也正在考虑 LDT 的监管事宜。11 FDA 已经注意到,IVD 制造商研发的检测方法与实验室自建检测方法相同却受到不同的监管,这本身就有待商榷。12
参考文献
- The NHS, National Pathology Program, https://www.england.nhs.uk/wp-content/uploads/2014/02/pathol-dig-first.pdf
- International Organization for Standardization (ISO) 15189. Schneider et al. Ann Lab Med. 2017 Sep
- ISO 15189:2012 Medical laboratories - Requirements for quality and competence. Westgard QC. Pereira, P. (February 2017).
- Medical biology in the face of the evolution of health care needs. Académie Nationale de Pharmacie. 2018
- Laboratoires de biologie médicale : analyse des coûts liés à l’accréditation selon la norme EN ISO 15189. Syndicat National des Médecins Biologistes. Mai 2011. http://www.bioprat.com/modules/upload/upload/coutsaccreditation.pdf
- SH-GTA 04 technical guide, https://tools.cofrac.fr/documentation/SH-GTA-04
- ISO 5725-4:1994. Accuracy (trueness and precision) of measurement methods and results -- Part 4, https://www.iso.org/fr/standard/11836.html
- Flow cytometry and the stability of phycoerythrin-tandem dye conjugates. R. Hulspas et al. Cytometry Part A. 2009
- SH FORM 43, COFRAC. https://tools.cofrac.fr/fr/documentation/index.php?fol_id=64
- Accreditation of Flow Cytometry in Europe. Sack et al. Cytometry Part B (Clinical Cytometry) 84B:135–142 (2013)
- FDA Notification and Medical Device Reporting for Laboratory Developed Tests (LDTs), Draft Guidance Document, US FDA, 2014 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-notification-and-medical-device-reporting-laboratory-developed-tests-ldts
- Discussion Paper on Laboratory Developed Tests (LDTs), US FDA, 2017 https://www.fda.gov/media/102367/download
