QA / QC放行检验

自动化生物制药质量控制以降低成本并提高数据完整性
阅读更多内容前言
生物制药药品的生产过程必须严格遵循质量法规和指南。GMP环境的主要侧重点是遵循21 CFR第11部分的数据完整性指南。GMP生产设施必须满足许多标准,而创建生产过程的审计跟踪是满足21 CFR第11部分标准的必要环节。FDA已发布数据完整性标准,简称ALCOA+原则。要求实验数据必须具备:
- 可溯源性,可与执行测量的仪器和用户关联。
- 易读性,以清晰、标准的格式呈现。
- 同步性,电子记录在生成数据时同步创建。
- 原始性,数据非转录或影印。
- 准确性,无更改且避免人工计算或手动数据录入。
- +, 确保记录仪停止运行后,数据仍完整可用。
生物制药生产工作流的每一个步骤,均需遵循以上指南,是避免成本高昂流程审计的关键。贝克曼库尔特生命科学公司分析仪,提供全面集成和自动化解决方案,为确保该工作流的每一步均符合ALCOA+原则保驾护航。
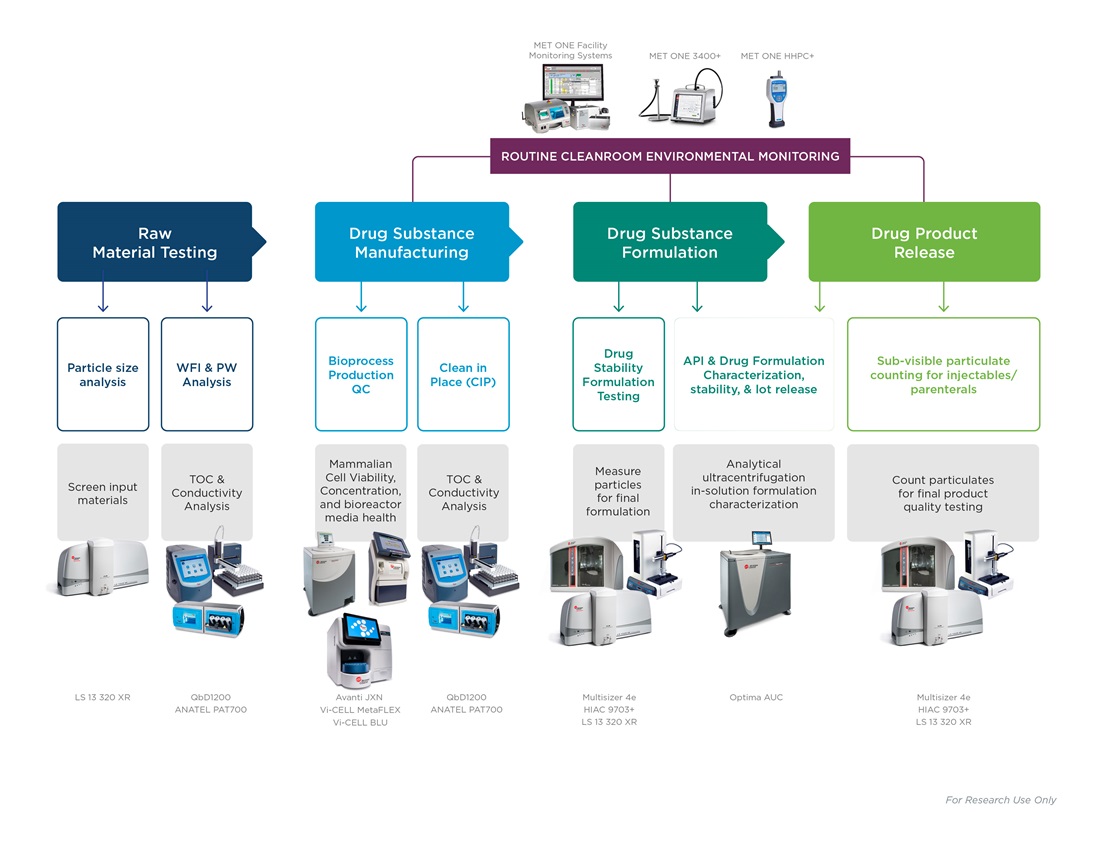
水质监测
在药物生产工作流和设施的常规就地清洗方案中,纯净水和注射用水的可用性是一个先决条件。水是生产中最重要的原材料。生产工作流的各个阶段所使用的进水,都必须经过总有机碳和电导率测定,范围包括:
- 细胞培养基
- 就地清洗和冲洗
- 最终制剂注射用水
进水中的任何潜在污染物都有可能危及整个下游工作流。因此,建议部署用于设施再循环回路的在线设备,持续检测水质,也可通过用于特定程序或质量测定实验室分析仪,在单个采样点进行水质检测。
环境监测
制药生产的空气质量条件受严格监管,需在洁净室和专门设计的环境中进行,空气中几乎没有颗粒物污染。必须不断评估生产空间和设施中的空气悬浮颗粒物,特别是来自于人体上的微生物。监测设备往往过于笨重,而且会发生人为错误或操作错误等意外情况。配备具有可根据预编程SOP自行配置并监测采样点功能的便携式空气计数器,有助于洁净室始终保持合规性。
原材料检验
评估生产用起始原料和辅料的一致性和适用性。干粉、悬浮液或乳化颗粒物在生产流程开始前即受到严格的质量控制。原料颗粒分布数据必须精确无误,才能确保最终产品的一致性,确保赋予患者有效的活性成分。自动化合格/未合格结果计算、预加载、按钮式SOP,助力用户轻松确定原材料的适用性。
生物生产
药品活性成分可由生物反应器内生长的细胞进行表达。必须密切监测培养条件和培养基,因为营养失衡和培养物成熟可能会影响产物得率。在指数生长期之后,或积累了过多生物量后,培养物开始凋亡,影响后续产量。细胞计数、活率和存活状态都是衡量生物生产的关键参数。
药物配方
通过细胞培养获得的活性成分与稳定剂或防腐剂等辅料混合,生成适用于患者的稳定溶液。在这一步骤中检测颗粒物或蛋白质聚集,对于确保不同批次和不同生产地点的产品一致性至为关键。
最终检验
最终制剂放行运输前,需根据当地药典指南(如USP <787>)评估颗粒污染和总体质量。注射用药品必须进行可见颗粒和亚可见颗粒的浓度和体积评估。治疗性蛋白质颗粒很小,不会影响检验结果;但,如果存在蛋白质聚集物,表明产品质量差。最终产品只有通过所有的质量检查后,方可放行药品运输。
结论
如何有效地遵守药品生产和质量控制的法规和指南颇具挑战。贝克曼库尔特生命科学公司为您提供各类先进仪器,助力企业在准确进行质量控制测量的同时,同步创建数据并安全导出原始电子记录。生产设施需长期生成和管理数据,依托贝克曼库尔特生命科学公司的各类分析仪,便可确保电子数据跟踪稳健可靠。
