Automated Cell Transfection and Reporter Gene Assay
Christophe Jung, Scientist
Peter Bandilla, Automation Specialist
Zhan Qi, PhD Student
Ulrich Unnerstall, Scientist
Ulrike Gaul, Principal Investigator
Department of Biochemistry, BioSysM / Gene Center, Ludwig-Maximilians-Universität München, Butenandtstr. 1, 81377
München, Germany
Abstract
Reporter gene assays have greatly helped our understanding of gene regulation and signal transduction in vitro and in vivo. They are widely used in molecular and cell biology studies, as well as in biomedical and pharmaceutical research and development. A typical reporter gene assay includes cell transfection of a reporter gene sequence cloned into an expression vector, and the determination of the gene expression level by measuring the reporter activity. We demonstrate here how a reporter gene assay can be automated using two Biomek NXP workstations, yielding accurate and highly reproducible measurements of reporter activity.
Introduction
Reporter gene assays are used to study gene expression and regulation. The most common applications include the testing of cis-regulatory sequences such as enhancers and promoters, functional analysis of promoter single nucleotide polymorphisms, studying signaling pathways, and drug discovery. Chemiluminescent reporters often outperform fluorescent, colorimetric or radioactive reporters due to their inherent sensitivity, large dynamic range and ease of set up. In particular, the low background light inherent in luminescence results in a better signal-to-noise ratio (SNR). Such high sensitivity is the prerequisite for most High-Throughput Screening (HTS) methods, which rely on the use of high-density plates to rapidly measure a single parameter across a large number of samples.
In a typical assay, cells are transfected with a vector that contains the sequence of interest cloned upstream of a luminescent reporter, such as Firefly or Renilla luciferase enzymes. In transient transfection experiments, an internal control with constitutive expression is often added, either as part of the same plasmid or co-transfected on a separate plasmid, in order to normalize for transfection efficiency, cell viability and sample handling. This system is known as the Dual Luciferase Reporter (DLR) assay. The activities of Firefly and Renilla luciferases are measured sequentially from a single sample, based on the different substrate specificities of the two luciferases, which provides a convenient means to measure both test and control reporter activities.
We sought to design a reporter gene assay based on the DLR system and with the following requirements:
- The assay should be fully automated (except for cell culture).
- Gene expression should be measured accurately and reproducibly for expression levels over multiple orders of magnitude.
- To ensure cost-effectiveness, reagent volumes should be minimized without impacting data quality.
We present here an automated protocol that fulfills these demanding requirements by using two Biomek NXP workstations (Figure 1A) for transient transfection and for the DLR assay of synthetic promoter variants. We will show that, besides increasing throughput, using automation leads to reduced variability of measurements over a dynamic range of up to five orders of magnitude.
Materials and Methods
Automation
Two Biomek NXP workstations, one with a 96-channel head and one with Span-8 pipettors, connected with a servo-shuttle, are used for all liquid transfer steps (Figure 1A). In addition, the robotic system is equipped with orbital shakers and a microplate reader (SpectraMax Paradigm, Molecular Devices) served by the Span-8 gripper. This configuration made it possible to fully automate the assay, from cell transfection to luminescence readout.
Vector system and cloning
All promoter constructs were cloned in the pGL4 Vector (Promega), which maximizes expression of Firefly and Renilla luciferases, while minimizing cryptic transcription factor binding sites. Firefly reporter was used to measure expression of test promoter variants, and Renilla was used for the control promoter (Drosophila pTRAN promoter). The GFP expression vector used to visualize transfected cells was a gift from K. Förstermann (Gene Center, Munich).
Cell culture and reagents
Drosophila Schneider 2 (S2) cells were cultured in serum-free Express V Medium (Invitrogen), supplemented with L-Glutamine (9%). 40,000 cells/well (each containing 110 μL of medium) were plated in 96-well plates (Corning) at passage 18. The cells were incubated within a humidified incubator at 25°C. Before transfection, Fugene HD (Promega) transfection reagent (diluted 16.2X) was mixed with the plasmid DNA (total concentration of 25.3 ng/μL) in water in four 96-well plates (4titude). We determined the optimal mass ratio between test and control plasmids to be 8:1 with our vector systems. The addition of the transfection reagent/DNA solution to the cells was then performed using the 96-channel head.
Readout of the luminescence signal
After incubation for 24 hours, the volume of the cell culture medium was reduced to 80 μL to increase luciferase concentration after lysis, and the cells were detached from the plate bottom by shaking. Subsequently, 2 × 20 μL of the lysed cell extract from each well of four 96-well cell culture plates was immediately added to two separate 384-well plates (AlphaPlates light gray, Perkin Elmer). Importantly, the plates were dark-adapted for 10 minutes before reading to reduce plate luminescent background. To each well of the first plate, we added 10 μL of One-Glo solution (Promega) containing a lysis buffer and the substrate for Firefly luciferase. The plate was transferred to the microplate reader and left for 10 minutes before measuring Firefly luminescence. The procedure was repeated with the second plate using the Renilla-Glo solution (Promega) to measure Renilla luminescence (for discussion, see below).
Automated transfection
For gene reporter assays, cell transfection is typically performed transiently, i.e. without integrating the plasmid DNA into the host genome. A preferred method is chemical transfection, where reagents package the DNA plasmid in liposomes, allowing them to pass through the cell membrane. This easy-to-use method can produce high transfection efficiency, low cell toxicity, and reproducible results. High transfection efficiency is important for providing a high signal and good well-to-well reproducibility, thereby helping to minimize experimental variability. Although normalization with the control reporter significantly reduces variability, a homogeneous transfection over the plate further improves data quality and avoids creating outlier wells (e.g. wells in which the control signal is too low).
The main difficulties in automating the transfection procedure are the need for strict control of cell culture conditions (splitting, use of serum-free medium, monitoring of cell proliferation/apoptosis), and the hydrophobic, viscous nature of the transfection reagent. Because of its low stability in aqueous environments and its adsorption to plastic containers and pipette tips, the transfection reagent had to be pipetted without dilution and at low volumes (2.3 μL). We used the Span-8 pipettors with P50 pipetting tips for this step. In order to limit adsorption, the lipophilic reagent was transferred into individual borosilicate glass tubes in 96-well plate storage format (Figure 1B). Eight tubes were used in parallel to optimize pipetting time and limit adsorption onto the tip surface. This procedure led to high transfection efficiency (50-60%), with well-to-well variability below 20% in a 96-well cell culture plate format (Figure 1C).
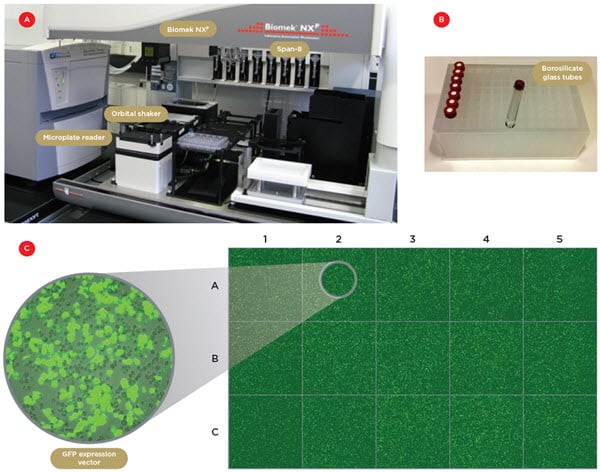
Figure 1. Robotic system and automated transfection. A) One of the two Biomek NXP workstations with a Span-8 pipettor, an orbital shaker and a microplate reader. B) The borosilicate glass tubes used to contain the lipophilic transfection reagent. C) Confocal tiled images of 15 individual wells of a 96-wellplate containing S2 cells transfected with a GFP expression vector. The transfection efficiency was 50-60% and the well-to-well variability below 20%.
Automated DLR Assay
To save time and reduce the required volume of the substrate reagents of the luciferase enzymes, it was highly desirable to perform the DLR assay in a 384-well plate format. While transfer of the cell culture and transfection protocol from 96-well to 384-well plates was not feasible, we managed to obtain similar performance for the DLR assay in the two different plate formats. We therefore proceeded by transferring our samples from 96-well to 384-well plates for the luciferase readout (Figure 2A). This enabled us to reduce the time required by the DLR assay, and to reduce the volume of substrate reagent by a factor of 6.
During the optimization process, we tested different plates, vector systems, luciferases and control promoters in order to optimize the signal-to-noise ratio (SNR). It turned out that the quality of the 384-well plates used for the readout is essential for maximizing the SNR and minimizing crosstalk of luminescent light between adjacent wells. We found that dark-adapted light gray plates gave the best results, with a crosstalk < 0.05%.
Another critical parameter is the delivery method for the two luciferase substrate reagents. We originally tested a widely used DLR kit (Dual-Glo, Promega), which provides the substrate for Firefly luciferase (driven by the test promoter), followed by the Renilla luciferase (driven by the control promoter). With this kit, the substrate for Firefly is added first, and the corresponding luminescence signal is measured. The reaction is then quenched, and the Renilla luciferase reaction initiated simultaneously by adding a second reagent to the same sample. However, we discovered two different problems when using this approach:
First, although the crosstalk value of 0.05% would be negligible for most applications, it represents a significant source of error in our experimental design, where extremely strong and weak promoters can occur side by side in neighboring wells, with a dynamic range exceeding three orders of magnitude.
Second, we found that the quencher added before the measurement of the Renilla signal was only partially quenching the Firefly signal for the strongest promoters. This resulted in overestimated Renilla signals, leading to erroneous normalization of the data.
We successfully solved both issues by modifying the assay pipeline. The cells were first lysed in medium, and then split, using the 96-channel head, into two different light gray plates for separate readout by adding Firefly substrate to one plate and Renilla to the other (Figure 2B). We verified that this procedure adds no variability to the normalized data, compared to sequential activity measurements using the same solutions. This demonstrates the high pipetting accuracy of the multichannel head. In this procedure, no quencher needs to be added before assaying the Renilla signal, which leads to a twofold increase in Renilla luminescence signal. Moreover, to address the cross-talk issue, we performed two readouts of the Firefly plate: the first readout after adding the substrate (post-incubation), and then a second measurement after pipetting out the high-signal solutions of strong promoters and replacing them with another quencher (Nile Blue) (Figure 2C). This procedure completely eliminated any crosstalk. To normalize for the slight loss in luminescence signal between the two readouts, as well as for normalization between different runs, different control promoters are systematically used as plate controls.
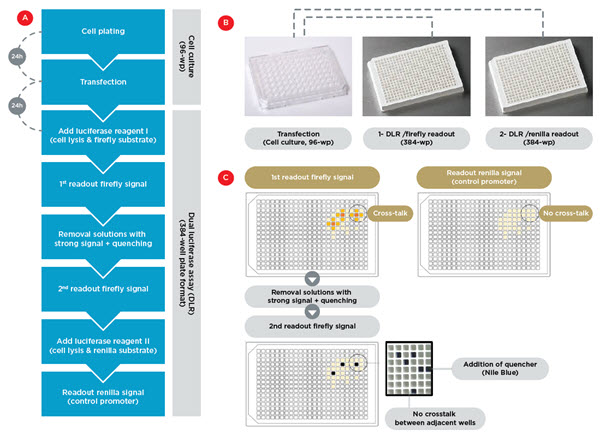
Figure 2. Assay workflow. A) Workflow for automated transfection in 96-wellplate format, followed by DLR assay in 384-well plate format. B) The transfection, lysis and cell detachement occurs in four cell culture 96-wellplates, followed by their splitting into two 384-well plates for separate readout of the firefly and renilla signals. C) Experimental strategy to eliminate crosstalk artifacts. Separating firefly and renilla readouts avoids crosstalk between the firefly and renilla luminescence light (the two upper panels). A second readout of the firefly signal after removal of the solution in wells with very strong signal eliminates the crosstalk between adjacent wells.
Assay performance
We found that, as expected, the use of a control promoter (Renilla signal) improves well-to-well reproducibility by a factor of 2 (Figures 3A and 3B), with a coefficient of variation (CV) between different wells < 10%. We also compared the normalized values of manual versus automated assays for promoter strengths distributed over as many as five orders of magnitude (Figure 3C and data not shown). The average CV decreased from 22% to 9%, demonstrating the significant advantages of automation.
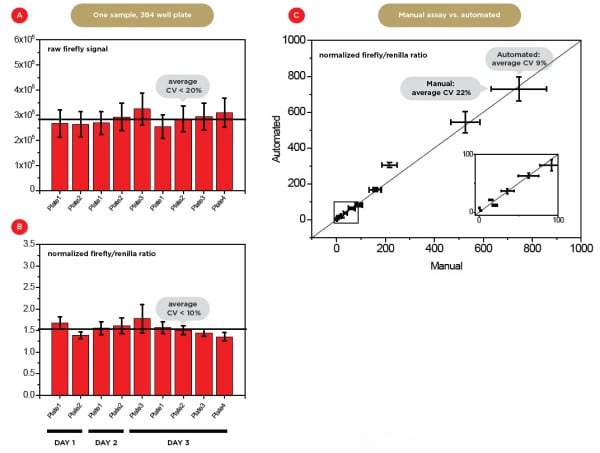
Figure 3. Assay reproducibility. A) Mean firefly raw luminescence signals for nine independent technical (same days) and biological (different days) replicates in 384-wp format. Each well arose from an independent transfection and contained the same experimental promoter with a weak expression level. B) Mean normalized expression level values. The average coefficient of variance (CV) decreases from ~20% to 10% after normalization. C) Comparison of normalized expression level values for manual versus automated sample preparation. The insert shows a magnification of the region highlighted with a black square. The expression levels of the 11 different experimental promoters tested covers a dynamic range of more than 3 orders of magnitude. The automated preparation leads to a much lower average CV (9%) for all promoters compared to manual preparation (22%).
Conclusion
In summary, we have shown that transferring a sensitive reporter gene assay to robotics using Biomek NXP workstations leads to substantially improved accuracy and reproducibility, in addition to higher throughput. Expression levels can be accurately determined over five orders of magnitude. The critical steps are to establish highly reproducible cell transfection, minimize luciferase substrate volume while maintaining high reproducibility, and eliminate artifacts due to the crosstalk between adjacent wells and between the two luciferase signals. The protocol was established using the DLR assay, but could easily be adapted to other common reporter systems such as fluorescence reporters.
Biomek Automated Workstations are not intended or validated for use in the diagnosis of disease or other conditions. Beckman Coulter Life Sciences genomic reagent kits are for research use only.
Helpful Links
-
阅读材料
-
应用手册
- 17-Marker, 18-Color Human Blood Phenotyping Made Easy with Flow Cytometry
- 21 CFR 第 11 部分关于在线 WFI 仪器的数据完整性要求
- 8011+ Reporting Standards Feature and Synopsis
- Achieving Compliant Batch Release – Sterile Parenteral Quality Control
- Air Particle Monitoring ISO 21501-4 Impact
- Automated 3D Cell Culture and Screening by Imaging and Flow Cytometry
- Automated Cell Plating and Growth Assays
- Automated Cell Transfection and Reporter Gene Assay
- Automated Cord Blood Cell Viability and Concentration Measurements Using the Vi‑CELL XR
- Automated Genomic Sample Prep RNAdvance
- Automated salt-assisted liquid-liquid extraction
- Automated Sample Preparation for the Monitoring of Pharmaceutical and Illicit Drugs by LC-MS/MS
- Automated XTT Assay for Cell Viability Analysis
- 自动化生物制药质量控制以降低成本并提高数据完整性
- Automating Cell-Based Processes
- Automating Cell Line Development
- The new Avanti J-15 Centrifuge Improves Sample Protection and Maximizes Sample Recovery
- Beer, Evaluation of Final Product and Filtration Efficiency
- Biomek Automated NGS Solutions Accelerate Genomic Research
- Biomek i-Series Automation of the Beckman Coulter GenFind V3 Blood and Serum DNA Isolation Kit
- Preparation and purification of carbon nanotubes using an ultracentrifuge and automatic dispensing apparatus, and analysis using an analytical centrifuge system
- Cell Counting Performance of Vi–Cell BLU Cell Viability Analyzer
- Viability Assessment of Cell Cultures Using the CytoFLEX
- Cell Line Development – Data Handling
- Cell Line Development – Limiting Dilution
- Cell Line Development – Selection and Enrichment
- 库尔特原理分析细胞
- Changes to GMP Force Cleanroom Re-Classifications
- Classifying a Small Cleanroom using the MET ONE HHPC 6+
- Clean Cabinet Air Particle Evaluation
- Recommended cleaning procedure for the exterior surface of the MET ONE 3400+
- 洁净室常规环境监测 —— FDA 关于 21 CFR Part 11 数据完整性要求
- 使用MET ONE 3400+ 进行 ISO 14644-3 洁净室自净时间测试
- Considerations of Cell Counting Analysis when using Different Types of Cells
- Consistent Cell Maintenance and Plating through Automation
- Control Standards and Method Recommendations for the LS 13 320 XR
- Counting Efficiency: MET ONE Air Particle Counters and Compliance to ISO-21501
- Critical Particle Size Distribution for Cement using Laser Diffraction
- Use Machine Learning Algorithms to Explore the Potential of Your High Dimensional Flow Cytometry Data Example of a 20–color Panel on CytoFLEX LX
- CytoFLEX
- Detecting and counting bacteria with the CytoFLEX research flow cytometer: II-Characterization of a variety of gram-positive bacteria
- Detecting Moisture in Hydraulic Fluid, Oil and Fuels
- Detection of foreign matter in plating solution using Multisizer4e
- Efficient kit-free nucleic acid isolation uses a combination of precipitation and centrifugation separation methods
- dsDNA Quantification with the Echo 525 Liquid Handler for Miniaturized Reaction Volumes, Reduced Sample Input, and Cost Savings
- Compensation Setup For High Content DURAClone Reagents
- Echo System-Enhanced SMART-Seq v2 for RNA Sequencing
- Efficient Factorial Optimization of Transfection Conditions
- European Pharmacopoeia EP 2.2.44 and Total Organic Carbon
- Flexible ELISA automation with the Biomek i5 Workstation
- Grading of nanocellulose using a centrifuge
- A method of grading nanoparticles using ultracentrifugation in order to determine the accurate particle diameter
- Grading of pigment ink and measurement of particle diameter using ultracentrifugation / dynamic light scattering
- HIAC Industrial – Our overview solution for fluid power testing for all applications
- A complete workflow for high-throughput isolation of DNA and RNA from FFPE samples using Formapure XL Total on the KingFisher™ Sample Purification System: an application for robust and scalable cancer research and biomarker discovery
- High-Throughput qPCR and RT-qPCR Workflows
- A Highly Consistent BCA Assay on Biomek i-Series
- A Highly Consistent Bradford Assay on Biomek i-Series
- A Highly Consistent Lowry Method on Biomek i-Series
- Highly Reproducible Automated Proteomics Sample Preparation on Biomek i-Series
- Cell Line Development – Hit Picking
- How to Use Violet Laser Side Scatter Detect Nanoparticle
- How Violet Side Scatter Enables Nanoparticle Detection
- HRLD Recommended Volume Setting
- Automating the Cell Line Development Workflow
- ICH Q2 – the Challenge of Measuring Total Organic Carbon in Modern Pharmaceutical Water Systems
- ICH Q2 – The Challenge of Measuring Total Organic Carbon in Modern Pharmaceutical Water Systems
- Importance of TOC measurement in WFI in light of European Pharmacopoeia change
- Integration of the Vi-CELL BLU Cell Viability Analyzer into the Sartorius Ambr® 250 High Throughput for automated determination of cell concentration and viability
- Issues with Testing Jet Fuels for Contamination
- Linearity of BSA Using Absorbance & Interference Optics
- Long Life Lasers
- LS 13 320 XR: Sample Preparation - How to measure success
- Particle Size Analysis Simple, Effective and Precise
- Beckman’s LS 13 320 XR Vs. Malvern Mastersizer
- Using Machine Learning Algorithms to Provide Deep Insights into Cellular Subset Composition
- Flow Cytometric Analysis of auto-fluorescent cells found in the marine demosponge Clathria prolifera
- MET ONE Sensor Verification
- Metal colloid purification and concentration using ultracentrifugation
- Separation and purification of metal nanorods using density gradient centrifugation
- High-throughput Miniaturization of Cytochrome P450 Time-dependent Inhibition Screening Using the Echo 525 Liquid Handler
- Miniaturization and Rapid Processing of TXTL Reactions Using Acoustic Liquid Handling
- Miniaturized Enzymatic Assays with Glycerol
- Miniaturized and High-Throughput Metabolic Stability Assay Enabled by the Echo Liquid Handler
- Miniaturized Multi-Piece DNA Assembly Using the Echo 525 Liquid Handler
- Minimal Sample to Sample Carry Over with the HIAC 8011+
- Minimizing process variability in the manufacturing of bottled drinking water
- Modern Trends in Non‐Viable Particle Monitoring during Aseptic Processing
- Particle diameter measurement of a nanoparticle composite - Using density gradient ultracentrifugation and dynamic light scattering
- Identification of Circulating Myeloid Cell Populations in NLRP3 Null Mice
- What to do now that ACFTD is discontinued
- Optimizing the HIAC 8011+ Particle Counter for Analyzing Viscous Fluids
- Optimizing the Multisizer 4e Coutler Counter for use with Small Apertures
- Optimizing Workflow Efficiency of Cleanroom Routine Environmental Monitoring
- Particle Counting in Mining Applications
- Particle testing in cleanroom high-pressure gas lines to ISO 14644 made easy with the MET ONE 3400 gas calibrations
- Pharma Manufacturing Environmental Monitoring
- Pharma Manufacturing Paperless Monitoring
- Analysis of plant genome sizes using flow cytometry: a case study demonstrating dynamic range and measurement linearity
- Flow Cytometric Approach to Probiotic Cell Counting and Analysis
- Protein purification workflow
- Calibrating the QbD1200 TOC Analyzer
- Detection Limit
- JP SDBS Validation
- USP System Suitability
- 符合《联邦法规 21 章》第 11 部分规定的质量控制电子记录
- Using the Coulter Principle to Quantify Particles in an Electrolytic Solution for Copper Acid Plating
- A Rapid Flow Cytometry Data Analysis Workflow Using Machine Learning- Assisted Analysis to Facilitate Identifying Treatment- Induced Changes
- Rapid Measurement of IgG Using Fluorescence Polarization
- Root Cause Investigations for Pharmaceutical Water Systems
- Full Automation of the SISCAPA® Workflow using a Biomek NXP Laboratory Automation Workstation
- Specification Comparison of Vi–CELL XR and Vi–CELL BLU
- Specifying Non-Viable Particle Monitoring for Aseptic Processing
- Switching from Oil Testing to Water and back using the HIAC 8011+ and HIAC PODS+
- 使用基于13色管的DURAClone干粉试剂在CytoFLEX流式细胞仪上进行人T细胞亚群的高级分析
- Comparative Performance Analysis of CHO and HEK Cells Using Vi-CELL BLU Analyzer and Roche Cedex® HiRes Analyzer
- USP 787 Small Volume Testing
- Validation of On-line Total Organic Carbon Analysers for Release Testing Using ICH Q2
- 采用 CytoFLEX 进行囊泡流式细胞术检测
- Vi-CELL BLU FAST Mode Option
- A fully automated plate-based optimization of fed-batch culture conditions for monoclonal antibody-producing CHO cell line
- A High-Throughput, Automated Screening Platform for IgG Quantification During Drug Discovery and Development
- The Valita Aggregation Pure assay: A rapid and accurate alternative for aggregation quantification of purified monoclonal antibodies
- Automated Research Flow Cytometry Workflow Using DURA Innovations Dry Reagent Technology with the *Biomek i7 Automated Workstation and *CytoFLEX LX Flow Cytometer
- Automating antibody titration using a CytoFLEX LX analyzer Integrated with a Biomek i7 Multichannel workstation and Cytobank streamlined data analysis
- Automated IDT Alt-R CRISPR/Cas9 Ribonucleoprotein Lipofection Using the Biomek i7 Hybrid Automated Workstation
- Automation of protein A ELISA Assays using Biomek i7 hybrid workstation
- Monitoring Plant Cell Cultures with BioLector and Multisizer 4e Instruments
- Monitoring E. coli Cultures with the BioLector and Multisizer 4e Instruments
- Monitoring Yeast Cultures with the BioLector and Multisizer 4e instruments
- Biomek i7 Hybrid Automated KAPA mRNA HyperPrep Workflow
- Cluster Count Analysis and Sample Preparation Considerations for the Vi-CELL BLU Cell Viability Analyzer
- Cultivation of suspended plant cells in the BioLector®
- How to use R to rewrite FCS files with different number of channels
- A new approach to nanoscale flow cytometry with the CytoFLEX nano analyzer
- CytoFLEX nano 纳米流式分析仪:纳米级流式细胞仪的前沿新技术
- 利用BioLector进行细胞死亡的测定
- Echo System-Enhanced SMART-Seq v4 for RNA Sequencing
- Filling MicroClime Environmental Lids
- Fully Automated Peptide Desalting for Liquid Chromatography–Tandem Mass Spectrometry Analysis Using Beckman Coulter Biomek i7 Hybrid Workstation
- A Simple Guide to Selecting the Right Handheld Particle Counter for Monitoring Controlled Environments
- High-throughput Miniaturization of Cytochrome P450 Time-dependent Inhibition Screening Using the Echo 525 Liquid Handler
- Host Cell Residual DNA Testing in Reduced Volume qPCR Reactions Using Acoustic Liquid Handling
- Linearity of the Vi-CELL BLU Cell Counter and Analyzer
- MET ONE 3400+ LDAP & Active Directory connection Guide
- 将 CytoFLEX S 流式细胞仪上设计的面板迁移至 CytoFLEX SRTl流式分选仪
- Miniaturization of an Epigenetic AlphaLISA Assay with the Echo Liquid Handler and the BMG LABTECH PHERAstar FS
- Miniaturization of Cytochrome P450 Time-dependent Inhibition Screening Using the Echo 555 Liquid Handler
- Miniaturized 16S rRNA Amplicon Sequencing with the Echo 525 Liquid Handler for Metagenomic and Microbiome Studies
- Miniaturized Enzo Life Sciences HDAC1 Fluor de Lys Assays Using an Echo Liquid Handler Integrated in an Access Laboratory Workstation
- Miniaturized EPIgeneous HTRF Assays Using the Echo Liquid Handler
- Miniaturized Gene Expression in as Little as 250 nL
- Miniaturized Genotyping Reactions Using the Echo Liquid Handler
- Nanoliter Scale High-Throughput Protein Crystallography Screening with the Echo Liquid Handler
- Nanoscale Sorting with the CytoFLEX SRT Cell Sorter
- Optimized NGS Library Preparation with Acoustic Liquid Handling
- Astrios和CytoFLEX SRT流式分选仪的孔板分选速度比较
- Preparation of Mouse Plasma Microsamples for LC-MS/MS Analysis Using the Echo Liquid Handler
- Purifying viral vector with VTi 90 rotor and CsCl DGUC
- Robust and High-Throughput SARS-CoV-2 Viral RNA Detection, Research, and Sequencing Using RNAdvance Viral and the OT-2 Platform
- 用CytoFLEX SRT细胞分选仪进行单细胞分选
- 用CytoFLEX SRT分选稀有E-SLAM造血干细胞及其后续培养
- Utilization of the MicroClime Environmental Lid to Reduce Edge Effects in a Cell-based Proliferation Assay
- Vertical Rotor Case Study with Adenovirus
- Variability Analysis of the Vi-CELL BLU Cell Viability Analyzer against 3 Automated Cell Counting Devices and the Manual Method
- Accurate enumeration of phytoplankton using FCM
- Accurately measures fine bubble size and particle count
- Adaptive Laboratory Evolution of Pseudomonas putida in the RoboLector
- Adjustment of the pH control settings in the BioLector® XT
- Aerobic cultivation of high-oxygen-demanding microorganisms in the BioLector XT microbioreactor
- An Analytical Revolution: Introducing the Next Generation Optima AUC
- Anaerobic cultivation processes of probiotic bacteria in the BioLector XT microbioreactor
- 使用 Multisizer 4e 库尔特颗粒计数及粒度分析仪监测贻贝/软体动物的繁殖
- Assay Assembly for Miniaturized Quantitative PCR in a 384-well Format Using the Echo Liquid Handler
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Automated Transfection Methods
- Automating a Linear Density Gradient for Purification of a Protein:Ligand Complex
- Automating Bradford Assays
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- The New Avanti J-15 Centrifuge Time Saving Deceleration Profile Improves Workflow Efficiency
- Avanti JXN Protein Purification Workflow
- Avoid the Pitfalls When Automating Cell Viability Counting for Biopharmaceutical Quality Control
- Biomek基因组样品制备自动化解决方案加速研究进程
- Biomek i-Series Automated AmpliSeq for Illumina® Library Prep Kit
- Biomek i-Series Automated Beckman Coulter Agencourt RNAdvance Blood Kit
- Biomek i-Series Automated Beckman Coulter Agencourt RNAdvance Cell
- Biomek i-Series Automated Beckman Coulter Agencourt SPRIselect for DNA Size Selection
- Biomek i-Series Automated IDT® xGen Hybridization Capture of DNA libraries on Biomek i7 Hybrid Genomics Workstation
- Biomek i-Series Automated Illumina Nextera DNA Flex Library Prep Kit
- Biomek i-Series Automated Illumina® Nextera XT DNA Library Prep Kit
- Biomek i-Series Automated Illumina TruSeq DNA PCR-Free Library Prep Kit
- Biomek i-Series Automated Illumina TruSeq® Nano DNA Library Prep Kit
- Biomek i-Series Automated Illumina TruSeq® Stranded mRNA Sample Preparation Kit Protocol
- Biomek i-Series Automated Illumina TruSeq® Stranded Total RNA Sample Preparation Kit Protocol
- Biomek i–Series Automated Illumina® TruSight Tumor 170 32 Sample Method
- Biomek i-Series Automated KAPA HyperPrep and HyperPlus Workflows
- Biomek i-Series Automated New England Biolabs NEBNext® Ultra II DNA Library Prep Kit
- Biomek i-Series Automated Promega Wizard MagneSil Tfx™ Plasmid Purification System
- Biomek i-Series Automated SurePlex PCR and VeriSeq PGS Library Prep for Illumina
- Biomek i-Series Automation of the DNAdvance Genomic DNA Isolation Kit
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Characterizing Insulin as a Biopharmaceutical Using Analytical Ultracentrifugation
- Comparing Data Quality & Optical Resolution of the Next Generation Optima AUC to the Proven ProteomeLab on a Model Protein System
- Control of Spheroid Size and Support for Productization
- Data-integrity-and-met-one-3400-plus-function-for-pharma
- Cultivation of Mammalian Cells in the Cydem VT System Bioreactor Module
- Cydem VT Automated Clone Screening System – Generating an Antibody Standard Curve
- Detection of Coarse Particles in Silica Causing Cracks in Semiconductor Encapsulants
- Determination of drug-resistant bacteria using Coulter counters
- Determination of Size and Concentration of Particles in Oils
- DO-controlled fed-batch cultivation in the RoboLector®
- Screening of yeast-based nutrients for E. coli-based recombinant protein production using the RoboLector Platform
- E. coli fed-batch cultivation using the BioLector® Pro
- Effective Miniaturization of Illumina Nextera XT Library Prep for Multiplexed Whole Genome Sequencing and Microbiome Applications
- Efficient clone screening with increased process control and integrated cell health and titer measurements with the Cydem VT Automated Clone Screening System
- Enhancing Vaccine Development and Production
- Enumeration And Size Distribution Of Yeast Cells In The Brewing Industry
- Evaluation of Instrument to Instrument Performance of the Vi-CELL BLU Cell Viability Analyzer
- Exosome-Depleted FBS Using Beckman Coulter Centrifugation: The cost-effective, Consistent choice
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Get Control in GMP Environments
- Getting Started with Kaluza: Data Scaling and Compensation Adjustment
- Getting Started with Kaluza: Parameters
- g-Max: Added Capabilities to Beckman Coulter's versatile Ultracentrifuge Line
- High throughput cultivation of the cellulolytic fungus Trichoderma reesei in the BioLector®
- High-throughput IgG quantitation platform for clone screening during drug discovery and development
- Illumina Nextera Flex for Enrichment on the Biomek i7 Hybrid Genomics Workstation
- Improved data quality of plate-based IgG quantification using Spark®’s enhanced optics
- Increased throughput for IgG quantification using Valita Titer 384-well plates
- Temperature dependence of hydrodynamic radius of an intrinsically disordered protein measured in the Optima AUC analytical ultracentrifuge.
- Introducing the Cydem VT Automated Cell Culture System: A high-throughput platform for fast and reliable clone screening experiments
- Jurkat Cell Analyses Using the Vi-CELL BLU Cell Viability Analyzer
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Matching Cell Counts between Vi–CELL XR and Vi–CELL BLU
- 利用RoboLector提高谷氨酸棒状杆菌蛋白质产量的培养基优化研究
- Method for Determining Cell Type Parameter Adjustment to Match Legacy Vi CELL XR
- Miniaturized Sequencing Workflows for Microbiome and Metagenomic Studies
- CytoFLEX SRT 上的混合模式分选
- Mode of operation of optical sensors for dissolved oxygen and pH value
- Modular DNA Assembly of PIK3CA Using Acoustic Liquid Transfer in Nanoliter Volumes
- Multi-Wavelength Analytical Ultracentrifugation of Human Serum Albumin complexed with Porphyrin
- Nanoliter Scale DNA Assembly Utilizing the NEBuilder HiFi Cloning Kit with the Echo 525 Liquid Handler
- Low-pH profiling in µL-scale to optimize protein production in H. polymorpha using the BioLector
- Performance of the Valita Aggregation Pure assay vs HPLC-SEC
- BioLector XT微型生物反应器小球藻光营养培养
- Precision measurement of adipocyte size with Multisizer4e
- Principles of Continuous Flow Centrifugation
- Protocols for use of SuperNova v428 conjugated antibodies in a variety of flow cytometry applications
- Purifying High Quality Exosomes using Ultracentrifugation
- Quality Control of Anti-Blocking Powder Particle Size
- Rapid Rabbit IgG Quantification using the Valita Titer Assay
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Screening yeast extract to improve biomass production in acetic acid bacteria starter culture
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Unveiling the Hidden Signals: Overcoming Autofluorescence in Spectral Flow Cytometry Analysis
- Unlocking Insights: The Vital Role of Unmixing Algorithms in Spectral Flow Cytometry
- A Standardized, Automated Approach For Exosome Isolation And Characterization Using Beckman Coulter Instrumentation
- Streamlined Synthetic Biology with Acoustic Liquid Handling
- SWOFF The unrecognized yet indispensable sibling of FMO
- The scattered light signal: Calibration of biomass
- Using k-Factor to Compare Rotor Efficiency
- Vaporized Hydrogen Peroxide Decontamination of Vi–CELL BLU Instrument
- Automating the Valita Titer IgG Quantification Assay on a Biomek i-Series Liquid Handling System
- Vi-CELL BLU 符合 21 CFRPart 11的法规要求
- Viral Vector Purification with Ultracentrifugation
- Analytical Ultracentrifugation (AUC) for Characterization of Lipid Nanoparticles (LNPs): A Comprehensive Review
- Leveraging Analytical Ultracentrifugation for Comprehensive Characterization of Lipid Nanoparticles in Drug Delivery Systems
- Whole Genome Sequencing of Microbial Communities for Scaling Microbiome and Metagenomic Studies Using the Echo 525 Liquid Handler and CosmosID
-
彩页
- Access Single Robot System——合成生物学工作流利器
- Automated Solutions for Cell Line Development
- Automated Solutions for ELISA
- Echo Acoustic Liquid Handling for Synthetic Biology
- HIAC 8011+ Liquid Particle Counting Systems
- HIAC 9703+ Sub-Visible Particulate Testing
- LS 13 320 XR - Laser Diffraction Particle Size Analyzer
- ValitaTiter IgG定量试剂盒彩页下载
-
案例分析
- Adenoviral Vectors Preparation
- Algae Biofuel Production
- Antibody and Media Development
- Autophagy
- B Cell Research
- Basic Research on Reproductive Biology
- Cardiovascular Disease Research
- Cell Marker Analysis
- Choosing a Tabletop Centrifuge
- Collagen Disease Treatment
- Controlling Immune Response
- Creating Therapeutic Agents
- DNA Extraction from FFPE Tissue
- English Safety Seminar
- Equipment Management
- Exosome Purification Separation
- Fast, Cost-Effective and High-Throughput Solutions for DNA Assembly
- Future of Fishing Immune Research
- Hematopoietic Tumor Cells
- High-throughput next-generation DNA sequencing of SARS-CoV-2 enabled by the Echo 525 Liquid Handler
- Hiroshima Genbaku HP Hematopoietic Tumor Testing
- iPS Cell Research
- Leveraging acoustic and tip-based liquid handling to increase throughput of SARS-CoV-2 genome sequencing
- Membrane Protein Purification X Ray Crystallography
- Organelles Simple Fractionation
- Particle Interaction
- Quality evaluation of gene therapy vector
- Retinal Cell Regeneration
- Sedimentary Geology
- Severe Liver Disease Treatment
- Tierra Biosciences reveals major molecular discovery
- Treating Cirrhosis
- University Equipment Management
- Fundamentals of Ultracentrifugal Virus Purification
- 产品目录
- 单页
-
专家访谈
- Background and Current Status of the Introduction of Flow Cytometers
- Benefits-of-the-coulter-principle-in-the-manufacturing-for-ips-cell-derived-natural-killer-cells
- Central Diagnosis in the Treatment of Childhood Leukemia 1
- Central Diagnosis in the Treatment of Childhood Leukemia 2
- Challenges-in-viability-cell-counting
- Contribution of Cytobank to 1-cell analysis of the cancer microenvironment
- Development of technology for social implementation of synthetic biology
- Flow Cytometry Testing in Hospital Laboratories
- Fundamentals of Ultracentrifugal Virus Purification
- The MET ONE 3400+ Automates Routine Environmental Monitoring for GMP Cleanroom Compliance
- Tumor Suppressor Gene p53 research and DNA Cleanup Process
- Fundamentals of Ultracentrifugal Virus Purification
- Dr Yabui UCF Lecture
-
主题报告
- Applications of Ultracentrifugation in Purification and Characterization of Biomolecules
- Automating Genomic DNA Extraction from Whole Blood and Serum with GenFind V3 on the Biomek i7 Hybrid Genomic Workstation
- ABRF 2019: Automated Genomic DNA Extraction from Large Volume Whole Blood
- Automated library preparation for the MCI Advantage Cancer Panel at Miami Cancer Institute utilizing the Beckman Coulter Biomek i5 Span-8 NGS Workstation
- Automating Cell Line Development for Biologics
- Cell-Line Engeneering
- Characterizing the Light-Scatter Sensitivity of the CytoFLEX Flow Cytometer
- AACR 2019: Isolation and Separation of DNA and RNA from a Single Tissue or Cell Culture Sample
- Mastering Cell Counting
- Preparing a CytoFLEX for Nanoscale Flow Cytometry
- A Prototype CytoFLEX for High-Sensitivity, Multiparametric Nanoparticle Analysis
- ABRF 2019: Simultaneous DNA and RNA Extraction from Formalin-Fixed Paraffin Embedded (FFPE) Tissue
- Quantification of AAV Capsid Loading Fractions: A Comparative Study
- Using Standardized Dry Antibody Panels for Flow Cytometry in Response to SARS-CoV2 Infection
- 产品说明书
- 实验步骤
-
白皮书
- Centrifugation is a complete workflow solution for protein purification and protein aggregation quantification
- AUC Insights - Analysis of Protein-Protein-Interactions by Analytical Ultracentrifugation
- A General Guide to Lipid Nanoparticles
- Addressing issues in purification and QC of Viral Vectors
- GMP Cleanrooms Classification and Routine Environmental Monitoring
- Purification of Biomolecules by DGUC
- AUC Insights - Assessing the quality of adeno-associated virus gene therapy vectors by sedimentation velocity analysis
- AUC Insights - Sample concentration in the Analytical Ultracentrifuge AUC and the relevance of AUC data for the mass of complexes, aggregation content and association constants
- Analyzing Biological Systems with Flow Cytometry
- 亚可见颗粒物检测新进展:USP <1788>的最新修订
- Changes to USP <643> Total Organic Carbon
- Characterization of RNAdvance Viral XP RNA Extraction Kit using AccuPlex™ SARS–CoV–2 Reference Material Kit
- CytoFLEX Platform Flow Cytometers with IR Laser Configurations: Considerations for Red Emitting Dyes
- Evaluation of the Analytical Performance of the AQUIOS CL Flow Cytometer in a Multi-Center Study
- Simultaneous Isolation and Parallel Analysis of gDNA and total RNA for Gene Therapy
- Hydraulic Particle Counter Sample Preparation
- Inactivation of COVID–19 Disease Virus SARS–CoV–2 with Beckman Coulter Viral RNA Extraction Lysis Buffers
- Tips for Cell Sorting
- IVD-R Annex I Global Safety and Performances Requirements
- Liquid Biopsy Cancer Biomarkers – Current Status, Future Directions
- MET ONE 3400+ IT Implementation Guide
- Reproducibility in Flow Cytometry
- SuperNova v428: New Bright Polymer Dye for Flow Cytometry
- SuperNova v428: New Bright Polymer Dye for Flow Cytometry
- Japan Document
-
应用手册
